P. Nimpiboon et al. / Process Biochemistry 46 (2011) 448–457
2.5. Purification of ˛-glucosidase
449
polyglucosylfructosides ranging from DP3 to DP8, with DP3 (mal-
tosylfructose, ␣-d-Glcp-(1 → 4)-␣-d-Glcp-(1 → 2)--d-Fruf) as the
main product [1,11]. In view of these findings, the search for an
enzyme with new properties for the synthesis of novel NDOs is of
prime concern.
Bacillus licheniformis is a Gram-positive, spore-forming soil bac-
terium that is commonly used in the biotechnology industry to
manufacture enzymes, antibiotics, biochemicals and consumer
products [12]. The thermotolerant B. licheniformis strain TH4-2 was
previously isolated from a soil sample in Thailand and screened
for the production of thermoactive levansucrase enzyme [13]. In
this work, we investigated the ␣-glucosidase activity of this bacte-
rial strain and explored its potential for use in transglucosylation
reactions for the synthesis of novel prebiotic OS.
Two liters of culture was prepared as above (Section 2.2), and then the bacterial
cells were removed by centrifugation at 3800 × g, 4 ◦C for 30 min. The supernatant
fraction was subjected to 30–60% saturation ammonium sulfate precipitation. The
precipitate was then dissolved in 20 mM sodium acetate buffer (pH 6.0), dialyzed
and loaded onto a DEAE-cellulose column (1.5 cm × 27 cm) in the same buffer. The
column was run at a flow rate of 0.75 ml/min and the enzyme eluted by linearly
increasing the NaCl gradient from 0 to 0.3 M (250 + 250 ml), collecting 5 ml frac-
tions. The fractions positive for ␣-glucosidase (sucrose hydrolysis activity) were
pooled and concentrated by ultrafiltration, then further purified by loading on a
Sephadex G-100 column (1.9 cm × 90 cm) in 20 mM sodium acetate buffer with a
flow rate of 0.33 ml/min and collecting 2 ml fractions. The fractions positive for ␣-
glucosidase (sucrose hydrolysis activity) were pooled and concentrated as the final
purified preparation and used for further analysis.
2.6. Characterization of ˛-glucosidase
2. Materials and methods
For both SDS- and native-PAGE, 7.5% (w/v) resolving gels were used. Enzyme
purity was followed by native-PAGE with protein staining by Coomassie Blue R-250
and enzyme activity staining. Zymogram staining of the sucrose hydrolysis activ-
ity was performed using the 2, 3, 5-triphenyltetrazolium chloride (TTC) method
as described [16]. The molecular weight of the purified enzyme was estimated by
SDS-PAGE.
2.1. Chemicals
p-Nitrophenyl
␣-d-glucopyranoside
(pNPGlc),
p-nitrophenyl
-d-
glucopyranoside, o-nitrophenyl -d-galactopyranoside, d-fructose, d-glucose,
lactose, isomaltose, maltooligosaccharides (G2 to G7), melibiose, cellobiose,
palatinose, ␣-amylase from Aspergillus oryzae, glucoamylase from Aspergillus niger
and rat intestinal acetone powder were obtained from Sigma (USA). Lactulose
was from Fluka (Switzerland) and sucrose from Bio Basic Inc. (Canada). Raffinose
pentahydrate was purchased from Nacalai Tesque, Inc. (Japan). Glucose oxidase kit
was a product of Human Biochemical and Diagnostics mbH (Germany). Acetonitrile
was from LAB-SCAN Analytical Science (Thailand). Other chemicals and solvents
used were of analytical grade.
2.6.2. Effect of pH and temperature on sucrose hydrolysis
The effect of pH was determined at 45 ◦C. The buffers (all 20 mM) used were
sodium acetate (pH 5.0–6.0), phosphate (pH 6.0–8.0) and borate (pH 8.0–9.0). The
effect of temperature was determined at the above determined optimum pH with
the temperature varied from 30 ◦C to 60 ◦C. The reaction was performed by incu-
bation of 10% (w/v) sucrose with 0.2 unit/ml enzyme for 10 min at variable pH or
temperature.
2.2. Strains and culturing condition
2.6.3. Substrate specificity
o-nitrophenyl -d-galactopyranoside, each at 5 mM, was determined by measuring
the amount of nitrophenol released. The activity towards sucrose, isomaltose, mal-
tose, maltotriose, cellobiose, lactose, melibiose, lactulose, palatinose and raffinose
(all at 50 mM) was evaluated by measuring the amount of glucose released by the
glucose oxidase-peroxidase method (GOD-POD) [17] using a glucose oxidase-based
kit (Human Biochemical and Diagnostics mbH, Germany). The enzyme’s hydrolytic
activity towards starch, amylose and amylopectin as substrates, all at 1% (w/v),
was followed by measuring the amount of glucose released using the method of
Somogyi–Nelson [14].
B. licheniformis TH4-2, a thermotolerant bacteria previously isolated from a soil
sample in Thailand [13], was used in this study. The medium used for ␣-glucosidase
production consisted of 1% (w/v) beef extract, 1% (w/v) peptone, 0.4% (w/v) ovalbu-
min, 0.5% (w/v) NaCl and 5% (w/v) soluble cassava starch at a pH of 6.5, except where
modified as indicated. Cultivation was performed at 45 ◦C with shaking at 250 rpm
for 42 h.
2.3. Identification of the Bacillus species
Bacillus sp. TH4-2 was identified by (i) morphological analysis and standard
biochemical tests (API 20C AUX system) and (ii) comparison of sequence similarity
and phylogenetic analysis of the existing species in the GenBank database using a
partial fragment (1544 bp) of the 16S rRNA gene.
2.7. Determination of kinetic parameters
2.7.1. Determination of Km and Vmax for pNPGlc
Hydrolysis of pNPGlc substrate was varied in the concentration range of
0.02–5 mM. The reaction was incubated with enzyme (0.15 unit/ml, pNPGlc hydrol-
ysis activity) in 20 mM sodium acetate buffer (pH 6.0) at 45 ◦C for 10 min. The activity
was determined as described under Section 2.4.1.
2.4. Enzyme and protein assay
␣-Glucosidase activity was assayed by the hydrolysis of pNPGlc, with moni-
toring of the level of the released p-nitrophenol. Twenty-five microliters of 5 mM
pNPGlc was incubated with 5 l enzyme solution in 20 mM sodium acetate buffer
(pH 6.0) in a total volume of 100 l at 45 ◦C for 10 min. The reaction was stopped
by adding 200 l of 1 M sodium carbonate, and the absorbance at 405 nm was mea-
sured [1]. One unit was defined as the amount of enzyme that produced 1 mol of
p-nitrophenol/min.
2.7.2. Determination of Km and Vmax for sucrose and maltose
Sucrose substrate was varied in the concentration range of 2.5–100 mM while
maltose was varied in the concentration range of 5–400 mM. The reaction was
incubated with enzyme (0.2 unit/ml, sucrose hydrolysis activity) in 20 mM sodium
acetate buffer (pH 6.0)at 45 ◦C for 10 min. Theactivity wasdetermined bythe glucose
oxidase-peroxidase method as in Section 2.6.3.
The level of sucrose hydrolysis was determined by measuring the amount of
reducing sugar produced by Somogyi–Nelson’s method using glucose as standard
[14]. The reaction mixture, containing 500 l of 20% (w/v) sucrose in 20 mM sodium
acetate buffer (pH 6.0), and 10 l of enzyme in 490 l of the same buffer, was incu-
bated at 45 ◦C for 10 min, and then the absorbance at 520 nm was measured. One unit
was defined as the amount of enzyme that produced 1 mol of reducing sugar/min.
At the fixed concentration of 150 mM sucrose, melibiose acceptor was varied in
the range of 100–500 mM. The reaction was incubated with enzyme (0.5 unit/ml,
sucrose hydrolysis activity) in 20 mM sodium acetate buffer (pH 6.0) at 45 ◦C for
180 min. The transglucosylation activity was evaluated by analyzing the products
by HPLC as described in Section 2.8.
2.8. Transglucosylation reaction of ˛-glucosidase and analysis of the products
2.4.2. Transglucosylation activity
The transglucosylation activity of ␣-glucosidase was determined by incubating
25 l of the enzyme preparation (0.5 units) with 475 l of 5% (w/v) sucrose in the
absence or presence of 5% (w/v) of the indicated saccharide acceptors in 20 mM
sodium acetate buffer pH 6.0. The reaction was incubated for 24 h at 45 ◦C, and then
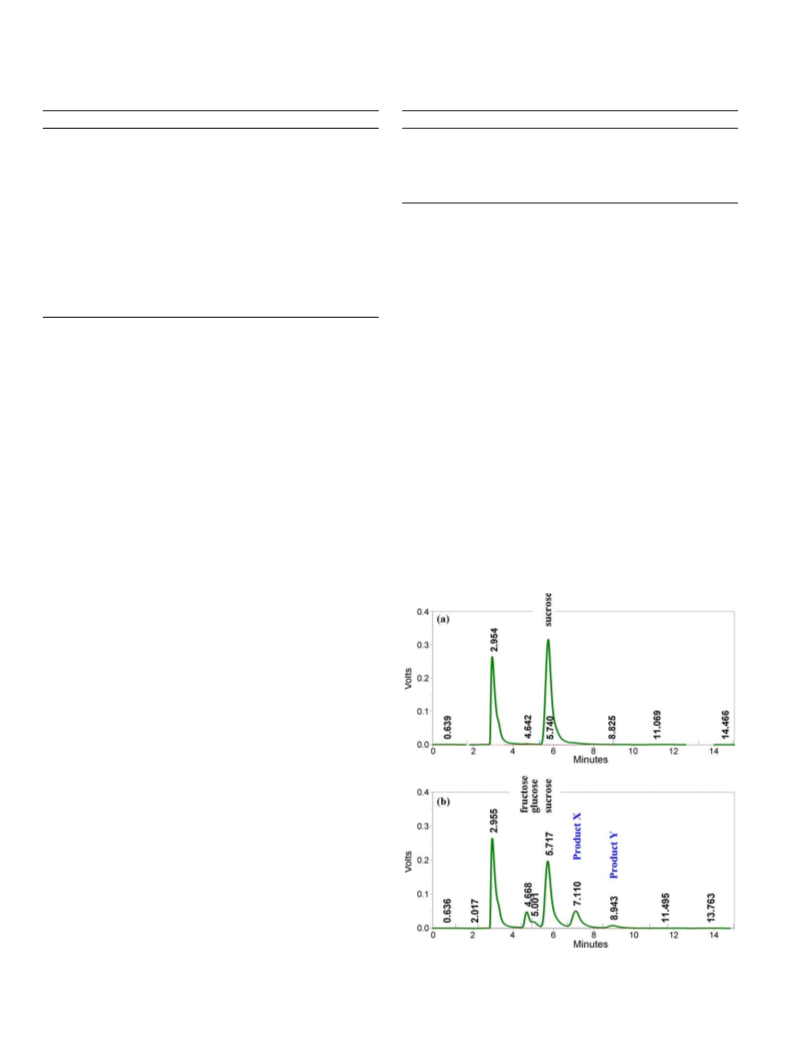
the transglucosylation activity was evaluated by analyzing the products by HPLC.
The transglucosylation reaction was followed by investigating the transfer prod-
ucts formed. The purified ␣-glucosidase (0.5 unit/ml, sucrose hydrolysis activity)
was incubated in 20 mM sodium acetate buffer, pH 6.0, with either 5% (w/v) sucrose
as a single substrate or with 5% (w/v) sucrose as the glucosyl donor and 5% (w/v)
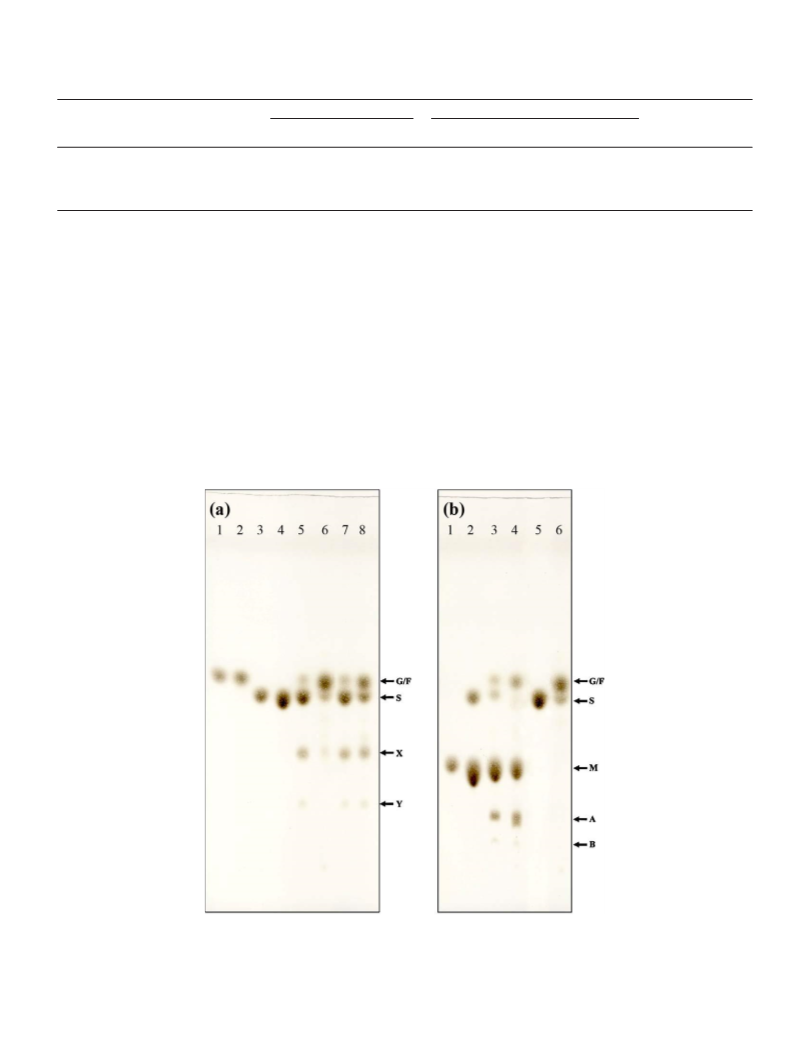
of one of the various acceptors (lactose, melibiose, cellobiose, raffinose, palatinose
and lactulose), as indicated, at 45 ◦C for 24 h. Then, the reaction mixture was boiled
and analyzed by TLC using a silica gel 60 plate (Merck), with a 7:1:2 (v/v/v) mixture
of n-propanol: ethyl acetate: water as the mobile phase solvent. After running, the
spots were detected by spraying with a 1:9 (v/v) mixture of concentrated sulfuric
acid: ethanol, followed by heating at 110 ◦C for 15 min.
2.4.3. Protein assay
Protein was quantified according to the Bradford method [15], using bovine
serum albumin as a standard. The fractions eluted from all chromatographic runs
were monitored for protein by measuring the absorbance at 280 nm.

 Nimpiboon, Pitchanan
Nimpiboon, Pitchanan