JOURNAL OF CHEMICAL RESEARCH 2007 413
Table 1 Continued
Entry Substrate
Product
Time/min
40
B.P./°C)
Yielda
Obsd
Lit
1
5
60–62/15c
192–194c
82
NOH
NOH
O
O
1
6
35
48–50b
47–51b
85
a
b
c
Isolated yields; melting point of compounds; boiling point under reduced pressure at mm Hg.
All the products were compared with authentic sample and gave satisfactory IR, NMR and MS data
The purity of the products which have different boiling points to their literature values, were checked by GC and TLC. R value and
f
retention time was matched with authentic samples. On distillation of these compounds at atmospheric pressure, it was observed
that yields of the products were reduced by 8–15%.
oxidation of the regenerated aldehydes. The method showed
chemo-selectivity. The oxidative deoximation of benzoin
(3 min). Helium was used as the carrier gas at a constant flow rate
of 1.2 ml/min. The samples were analysed in the splitless mode at
injection temperature of 250°C, EI source temperature 230°C and
quadrupole analyser at 150°C.
(entry 8) and the unsaturated oxime (cinnamaldoxime, entry
7
) proceeded without affecting the hydroxyl group and double
bond respectively. In order to examine the chemo-selectivity
of the method further, two sets of experiments were performed
under similar reaction conditions. In the first set of experiments,
an equimolar mixture of benzophenone oxime and styrene
were allowed to react with CC-2. The benzophenone oxime
under went chemo-selective deoximation giving 85%
benzophenone without any observable oxidation of styrene.
Similarly in another experiment, an equimolor mixture of
benzophenone oxime and benzyl alcohol, were allowed to
react with CC-2 at room temperature. The benzophenone
oxime underwent chemoselective oxidative deoximation
giving (85%) benzophenone, whereas benzyl alcohol was
recovered almost quantitatively. It indicated non-competitive
oxidation of oximes in the presence of hydroxyl group as well
as with a double bond.
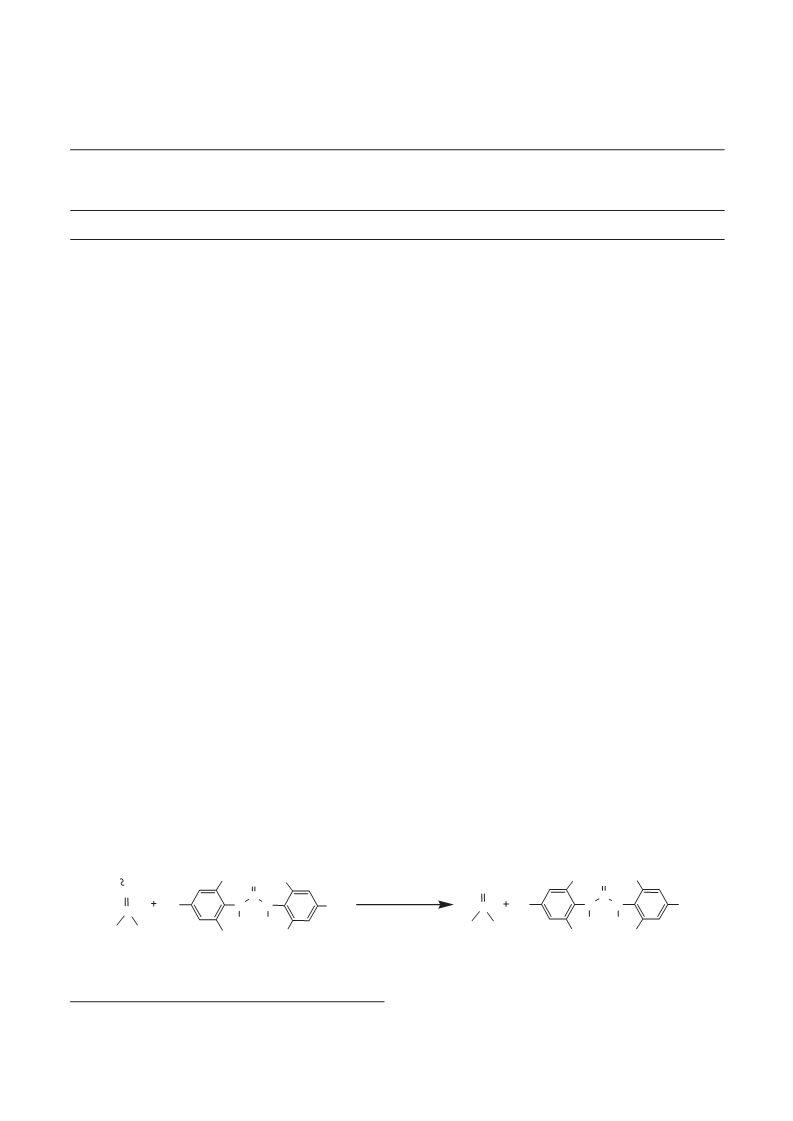
Typical experimental procedure
In a typical experimental procedure, a solution of benzophenone
oxime, 9.85 g (0.05 mol) in 50 ml acetonitrile was added slowly to
a suspension of CC-2, 12.2 g (0.025 mol) in 25 ml acetonitrile–H O
2
mixture (8:2) with stirring at room temperature. The progress of
the reaction was monitored by TLC. The completion of reaction
was also indicated by ceasing the precipitation of bis-(2,4,
6
-trichlorophenyl) urea. The reaction mixture was filtered by suction
and washed with acetonitrile (4 × 10 ml). The solvent was evaporated
and the residue was cooled under ice cold which gave crude solid
product followed by recrystallisation from diethyl ether to afford
pure crystals of benzophenone; yield: 7.74 g (85%), m.p. 47–48°C
(lit 46–48°C).
WethankDrR.Vijayaraghavan,Director,andProf.M.P.Kaushik,
Associate Director, DRDE, Gwalior, for their keen interest,
encouragement and fruitful discussion.
In conclusion, we describe a simple, mild, convenient and
chemo-selective method for the oxidative deoximation of
oximes by the use of CC-2. The hydroxyl and alkene groups
are not affected. Finally the dechlorinated product (4) can be
converted to (2) by rechlorination and could be reused several
times making it recyclable.
Received 21 April 2007; accepted 6 June 2007
Paper 07/4615 doi: 10.3184/030823407X218093
References
1
(a) T.W. Greene and P.G. Wuts, Protective Groups in Organic
Synthesis, 3rd edn., John Wiley & Sons, New York, 1999, pp. 355-359;
Experimental
(b) B.P. Bandgar, B.K. Lalita and J.L. Thote, Synth. Commun., 1997,
All the oximes were synthesised by reported methods. The chemicals
required for synthesis of oximes and CC-2 was purchased from
Aldrich Chemical Company USA. Solvents and other chemicals
such as pyridine, NaOH, hydroxylamine hydrochloride, acetic acid,
and acetonitrile were obtained from S.D. Fine Chemicals, Mumbai,
India.
2
7, 1149; (c) L.G. Donaruma and W. Heldt, J. Org. React., 1960, 11, 1;
(d) A.L. Bosch, P. Cruz, E. Diez-Barra, I. Loupy and F.Lanya, Synlett.,
1995, 1259.
E.H. Massey, B. Kitchell, L.D. Martin, K. Gerren and H.W. Murphy,
Tetrahedron. Lett., 1970, 11, 157.
2
3
4
5
R.V. Sterens, F.C.A. Gaeta and D. Lawrence, J. Am. Chem. Soc., 1983,
1
05, 7713.
A.K. Gupta, D.K. Dubey and M.P. Kaushik, Org. Prep. Proced. Int., 2005,
7, 294.
(a) R.H. Barry and W.H. Hartung, J.Org. Chem., 1947, 12, 460;
b) D.H.R. Barton, J.M. Beaton, L.E. Geller and M.M. Pechet, J. Am.
IR spectra were recorded on Bruker FT-IR spectrometer model
1
Tensor 27 as KBr disks. H NMR spectra were recorded on Bruker
3
DPXAvance FT- NMR in CDCl or DMSOd6 using tetramethylsilane
3
as an internal standard at 400 MHz. A Chemito GC model 1000
instrument was used with flame ionisation detector (FID). A capillary
column (30 m × 0.25 mm I.D-BP5) packed with 5% phenyl and 95%
dimethyl polysiloxane (SGE) coated on fused silica was employed.
The injection port and detector block were maintained at 280°C
and 260°C respectively and the column oven was at programmed
temperature profile started at 50°C, ramped up to 280°C at 25°C/
min. Nitrogen was used as a carrier gas (at a flow rate of 30 ml/min).
Air for FID was supplied at 300 ml/min and hydrogen at 30 ml/min.
In all analysis, 1ml sample were injected and peaks recorded on
computerized data acquisition station. The GC–MS analyses were
performed in EI (70 eV) in full scan mode with an Agilent 6890
GC equipped with a model 5973 mass selective detector (Agilent
Technologies, USA). An SGE BPX5 capillary column with 30 m
length × 0.32 mm internal diameter × 0.25 mm film thickness was
used at temperature program of 80°C (2 min)–20°C/min–280°C
(
Chem. Soc., 1961, 83, 4076; (c) D.H.R. Barton and J.M. Beaton, J. Am.
Chem. Soc., 1961, 83, 4053; (d) O. Touster, Org. React., 1953, 7, 327.
(a) G. Zhay, D. Yang, M. Chem and K. Cai, Synth. Commun., 1998, 28,
2221; (b) P.M. Bendale and B.M. Khadilkar, Tetrahedron Lett., 1998, 39,
5867 (c) G. Shang, D. Yang, M. Chem, Synth. Commun., 1998, 28, 3721;
(d) A. Boruah, B. Baruah, D. Prajapati and J.S. Sandhu, Tetrahedron Lett.,
6
7
1
3
997, 38, 4267; (e) D.S. Bose and P. Srinivas, Synth. Commun., 1997, 27,
835; (f) T. Shinada and K.Yoshihara, Tetrahedron Lett., 1995, 36, 6701.
(a) D.P. Curran, J.F. Brill and D.M. Rahiewio, J. Org. Chem., 1983, 49,
654; (b) C.G. Rao, A.S. Rashakrishna, B.B. Singh and S.P. Bhatnagar,
1
Synthesis, 1983, 803; (c) S.B. Shim, K. Kim and Y.H. Kim, Tetrahedron
Lett., 1987, 28, 645; (d) J.R. Meloney, R.E. Lyle, J.E. Saavedra
and G.G. Lyle, Synthesis, 1978, 212; (e) R. Joseph, A. Sudalai and
T. Ravindranathan, Tetrahedron, 1994, 35, 5493; (f) B. Bandgar,
L.B. Kunde and J.L. Thote, Synth.Commun. 1997, 27, 1149; (g) G.A. Olah,
Q. LIao, E.S. Lee and G.K. Surya Prakash, Syn. Lett., 1993, 427.
PAPER: 07/4615

 Gupta, Arvind K.
Gupta, Arvind K.