J. McNulty et al. / Bioorg. Med. Chem. Lett. 25 (2015) 4114–4117
4115
HS
SH
S
S
RS SR'
+
+
R-SH
+ R'SH
HO
OH
HO
OH
DTT
Scheme 1. Reduction of disulfide bridge or thiol protection mediated by the standard reagent DTT.1
HS
HO
SH
observed to be significantly more stable than that of TCEP or DTT.
(
HO CCH CH ) P
(HO-CH CH CH ) P
2 2 2 3
The half-life of DTT at pH 8.0 under these conditions was observed
2
2
2 3
OH
DL-DTT
to be approximately 4 h, in accord with literature values.2 While
TCEP
THPP
the relative stability of TCEP relative to DTT has been remarked
1
5b
upon before,
TCEP
literature data on the relative stability of
Scheme 2. Water soluble trialkylphosphines TCEP and THPP used in this study in
comparison to the standard reagent DL-DTT.
1
1,15a,b
15d
and THPP
are difficult to directly compare as they
have been performed at differing pH’s in various buffers. The direct
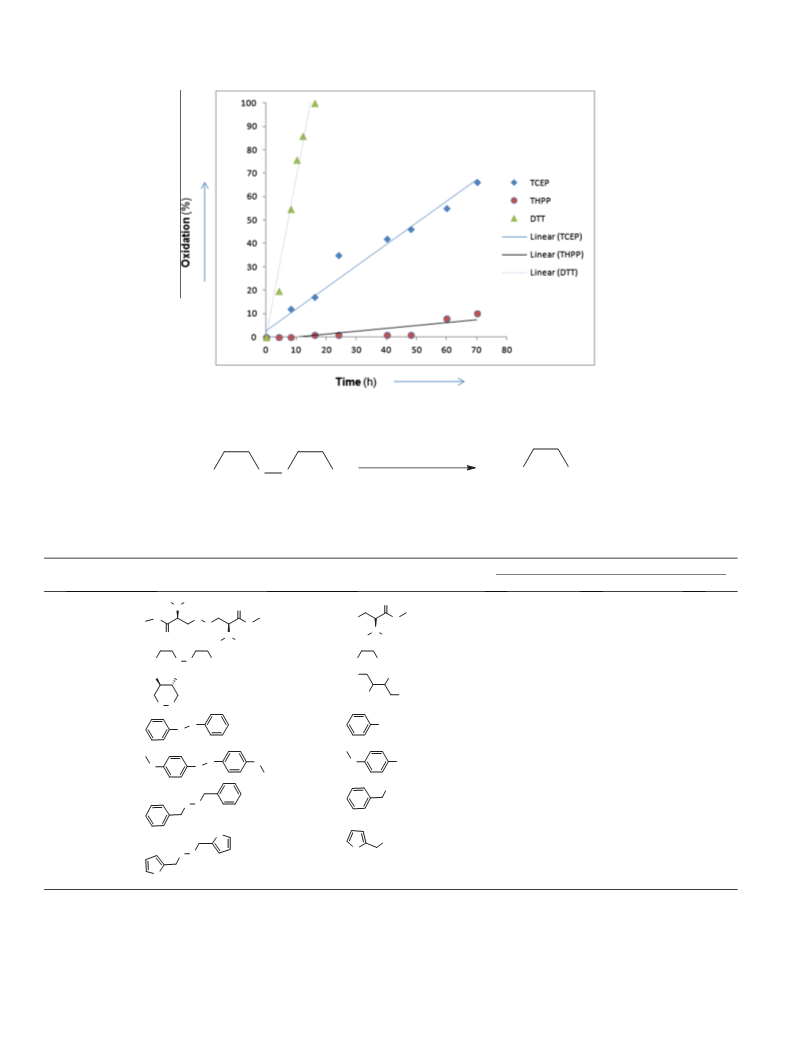
comparison reported here (Fig. 2) shows the exceptional stability
of THPP under physiological conditions, suffering only 10% oxida-
tion under these conditions over 72 h.
and, in conjunction with the Whitesides reagent, prompted the
present comparative investigation into the air-stability and reduc-
ing ability of THPP directly with TCEP and DTT in the reduction of
small-molecule disulfides. The results of this analysis and investi-
gation into the scope of small-molecule disulfide reduction medi-
ated by THPP are reported herein.
THPP was found to be soluble in water in all proportions, giving
rise to an odorless solution. The oxidative stability of aqueous solu-
tions of DTT, TCEP and THPP were compared at pH 4.50 (citrate
based buffer) and 8.00 [tris-(hydroxymethyl)-aminomethane-
In order to gauge the relative reducing power of TCEP and THPP,
the 2-hydroxyethyl disulfide 1b was prepared and experiments
were performed involving the conversion of this compound to
the corresponding thiol 2b (Scheme 3). The reduction of 1b proved
to be very fast at room temperature using THPP in comparison to
TCEP or DTT. The reduction of 1b using 1.1 equiv of THPP, TCEP
and DTT was followed in three separate side by side experiments
at pH 8.0 buffer. After 15 min, the reduction with THPP was com-
plete (82% isolated yield) while TCEP provided 25% and DTT 30%
isolated yields under these identical conditions.
In order to quantify the relative rates of reduction of 1b using
TCEP and THPP, direct competition experiments were performed
with a 1.0:1.0 solution of TCEP and THPP in aqueous citrate buffer
at pH 4.5 and 8.0. The addition of 0.67 equiv of the disulfide 1b
resulted in its rapid reduction to 2b. Analysis of the NMR spectra
showed formation of the phosphine oxides of THPP and TCEP in
the ratio of 1.00:1.25. Repetition of the same procedure using
Tris-buffer at pH 8.0 showed formation of phosphine oxides of
THPP and TCEP in the ration 1.50:1.00 indicating a reversal in the
relative rates of reduction and showing THPP to be the superior
reducing reagent at biologically relevant pH. Similarly, the reduc-
tion of the peptide-like disulfide 1a was shown to proceed 3.91
times faster with THPP relative to TCEP at pH 8.0 buffer.
2
CaCl based buffer] through the direct monitoring of aqueous solu-
tions of each by NMR analysis. The NMR spectra of THPP and TCEP
are complicated by the formation of the protonated phosphines
around pH 6–7. At pH 4.50 the phosphines are fully protonated
while at pH 8.00 the free base of each is clearly observed. Hence
3
1
the P NMR of THPP at pH 4.50 shows the phosphorus resonance
at d +15.8 ppm, corresponding to the protonated form (pK
a
1
5a
31
reported at 7.2).
At pH P 7, the P-resonance for the free base
is observed at À30 ppm, while the corresponding phosphine oxide
is observed at +60 ppm. The 31P NMR of TCEP at pH 4.50 similarly
demonstrated a resonance at d +17 ppm corresponding to the pro-
tonated TCEP species, a resonance at À26 ppm at pH P 7, corre-
sponding to TCEP free base and the oxide of TCEP at +56 ppm. In
case of DTT, progress of the auto-oxidation was conveniently fol-
1
lowed by H NMR. The methylene multiplet resonance for DTT
was observed at d 2.47 ppm, clearly distinguished from the corre-
sponding peak for the cyclic disulfide centered at d 2.41 ppm.
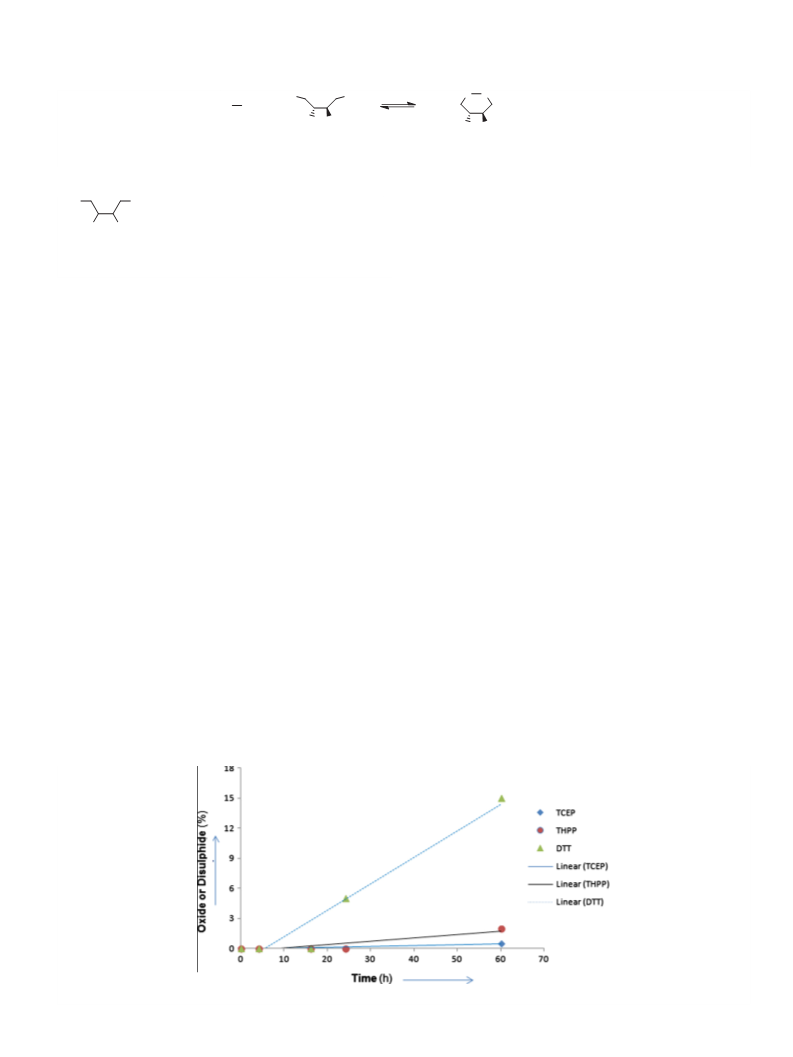
The results of these analyses are reported in Figures 1 and 2. At
pH 4.50 (Fig. 1) DTT demonstrated considerable auto-oxidation
after 2–3 days while both of the phosphines proved to be relatively
stable, most likely being protected in their salt forms. More impor-
tantly, at physiological pH (Fig. 2) aqueous solutions of THPP were
The scope of the use of THPP, TCP and DTT in reducing a group
of selected disulfides was next investigated, the results of which
are collected in Table 1. The disulfides 1a–g were prepared follow-
1
6,17
ing literature methods.
Structures were chosen to cover a
range of aryl, heterocyclic, benzylic and aliphatic disulfide func-
tionalities. As such, the polarity and solubility of these starting
Figure 1. Relative stability of aq solutions of DTT, TCEP and THPP to room temperature auto-oxidation at pH 4.50.

 McNulty, James
McNulty, James