ChemComm
Communication
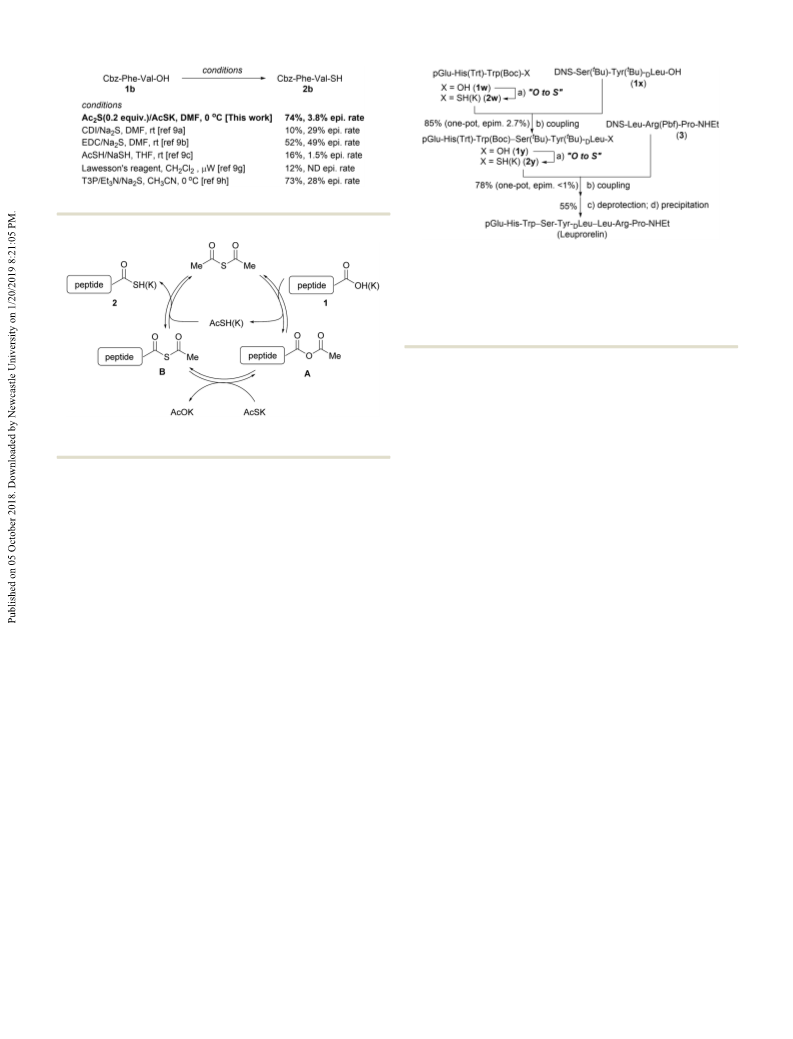
Scheme 2 Comparison to previously reported peptide thioacid syntheses.
Scheme 4 Application of the catalytic one-step transformation of pep-
tides into peptide thioacids to the iterative peptide–fragment-coupling
reaction for the synthesis of leuprorelin; pGlu = L-pyroglutamic acid; DNS =
2
,4-dinitrobenzenesulfonyl; HOObt = 3-hydroxy-1,2,3-benzotriazin-4-one.
(
4
a) AcSK (3 equiv.), Ac S (20 mol%), DMF, rt; (b) DNS–peptide (1x or 3,
2
i
3 2 2 3
equiv.), HOObt (1 equiv.), DMF, rt; (c) CF CO H/H O/Pr SiH (95/2.5/2.5), rt;
(
d) Et
2
O.
scope is broad; (4) the epimerization levels are very low; (5) the
reactions can be easily up-scaled (gram scale). The method can
also be efficiently combined with the thioacid-selective peptide-
bond-forming reaction developed by Crich, which was used to
carry out a convergent synthesis of leuprorelin. Up to now, the
scope of the current conditions is limited to side-chain protected
peptides, remaining to be improved in the future. Extensions of
Scheme 3 Plausible reaction mechanism for the catalytic one-step trans-
formation of peptides into peptide thioacids.
ꢀ
of the highly nucleophilic AcS is presumably a key factor to this method into the solid-phase synthesis of peptide thioacids
promote the O-to-S exchange at a rate faster than that of the and a more atom-economical coupling protocol are currently in
intramolecular cyclization that generates the configurationally progress.
labile oxazolone, which would cause epimerization.
To demonstrate the synthetic utility of this method for
the generation of peptide thioacids, a convergent synthesis of
Conflicts of interest
leuprorelin, a non-natural peptide drug comprising nine amino
acid residues, was performed via iterative peptide–fragment-
There are no conflicts to declare.
coupling reactions (Scheme 4). For that purpose, we selected Notes and references
the 2,4-dinitrobenzenesulfonyl (DNS)–amide–thioacid-coupling
1
(a) K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20,
22–128; (b) D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem.
Biol. Drug Des., 2013, 81, 136–147; (c) A. A. Kaspar and J. M. Reichert,
Drug Discovery Today, 2013, 18, 807–817; (d) P. Vlieghe, V. Lisowski,
J. Martinez and M. Khrestchatisky, Drug Discovery Today, 2010, 15,
8f,g
reaction developed by Crich,
given that the DNS amide is
1
14
easily prepared via a solid-phase synthesis and that there are no
restrictions regarding the amino-acid residues at the coupling
site. Leuprorelin was retrosynthetically divided into three peptide
fragments with protected side chains (1w, 1x, and 3), and these
peptide fragments were sequentially coupled from the N- to the
C-terminus. Thus, after conversion of 1w into 2w (or its potassium
salt), DNS amide 1x was added without isolation of 2w to produce
hexapeptide 1y in 85% yield. The addition of 3-hydroxy-1,2,3-
benzotriazin-4-one (HOObt) was crucial in order to increase the
40–56.
2 D. G. Brown and J. Bostr ¨o m, J. Med. Chem., 2016, 59, 4443–4458.
3
4
(a) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479;
b) H. Lundberg, F. Tinnis and H. Adolfsson, Chem. Soc. Rev., 2014,
3, 2714–2742; (c) R. M. de Figueiredo, J.-S. Suppo and J.-M.
(
4
Campagne, Chem. Rev., 2016, 116, 12029–12122.
(a) E. Bayer and M. Mutter, Nature, 1972, 237, 512–513; (b) L. A. Carpino,
S. Ghassemi, D. Lonescu, M. Ismail, D. Sadat-Aalaee, G. A. Truran,
E. M. E. Mansour, G. A. Siwruk, J. S. Eynon and B. Morgan, Org. Process
Res. Dev., 2003, 7, 28–37; (c) D. Takahashi and T. Yamamoto, Tetrahedron
Lett., 2012, 53, 1936–1939; (d) L. Andersson, L. Blomberg, M. Flegel,
L. Lepsa, B. Nilsson and M. Verlander, Biopolymers, 2000, 55, 227–250.
(a) S. B. Kent, Chem. Soc. Rev., 2009, 38, 338–351; (b) S. B. Kent,
Bioorg. Med. Chem., 2017, 25, 4926–4937.
1
5,16
yield and to suppress the epimerization level (2.7%).
The
second fragment coupling (2y + 3) also proceeded smoothly to
produce protected leuprorelin in 78% yield and o1% epimeriza-
tion level. A subsequent deprotection of the side chain, followed
by precipitation produced leuprorelin in 55% isolated yield.
In conclusion, we have developed a catalytic one-step synthesis
of peptide thioacids. The following features of this method are
noteworthy: (1) the generation of waste derived from protecting
groups and coupling reagents is minimal; (2) only inexpensive
and commercially available reagents are used; (3) the substrate
5
6
(a) L. Raibaut, N. Ollivier and O. Melnyk, Chem. Soc. Rev., 2012, 41,
7
2
001–7015; (b) L. R. Malins and R. J. Payne, Curr. Opin. Chem. Biol.,
014, 22, 70–78; (c) E. Saxon, C. R. Armstrong and C. R. Bertozzi, Org.
Lett., 2000, 2, 2141–2143; (d) B. L. Nilsson, L. L. Kiessling and
R. T. Raines, Org. Lett., 2000, 2, 1939–1941; (e) J. W. Bode, Acc.
Chem. Res., 2017, 50, 2104–2115; ( f ) S. Aimoto, Biopolymers, 1999,
51, 247–265; (g) J.-S. Zheng, S. Tang, Y.-C. Huang and L. Liu, Acc.
Chem. Res., 2013, 46, 2475–2484.
1
2224 | Chem. Commun., 2018, 54, 12222--12225
This journal is ©The Royal Society of Chemistry 2018

 Matsumoto, Takuya
Matsumoto, Takuya