Organic Letters
Letter
1B),9 we observed that the relative alcohol stereochemistry had
a significant effect on the reaction pathway. In using model
substrate 2-Me-cyclohexanol (2), cis-2 gave a clean rearrange-
ment to cyclic ether 3, whereas trans-2 led to significant
amounts of competitive ketone formation (4). We rationalized
this divergent reactivity through differences in the conforma-
tional equilibria of the two substrates. The trans-2 placed the
alcohol equatorial in the major conformer, resulting in an
intermediate iodate ester that could readily achieve the
necessary antiperiplanar orbital alignment with the α-hydrogen
(2-trans-OHeq), enabling facile oxidation. In contrast, the cis-2
was biased toward an axial alcohol conformer, wherein
significant steric clash with the 1,3-diaxial hydrogens inhibited
access to the necessary orbital alignment, thus leading to
rearranged products (2-cis-OHax). On the basis of this model,
we recognized that if the N-HVI reactivity could instead be
tuned to favor oxidation, this could lead to the first general
method for the chemoselective oxidation of equatorial over
axial alcohols. Furthermore, this would provide a rare example
of broad-scope alcohol oxidation with a λ3-iodane reagent as
the sole oxidant,13 providing practical advantages over the
current λ5-iodane oxidants.12,14,15
Herein we report the successful development of two
practical protocols for alcohol oxidation using simple
pyridine-ligated N-HVI, Py-HVI (5), and its utility in the
chemoselective oxidation of equatorial alcohols. The oxidation
can be performed either with isolated Py-HVI or via its in situ
generation, enabling a one-pot procedure from commercial
PhI(OAc)2 (Scheme 1C). The oxidation shows a broad
substrate scope and functional group tolerance. In cyclic
substrates, Py-HVI displays excellent selectivity for equatorial
alcohols in both conformationally flexible 1,2-substituted
cyclohexanols and rigid polyol scaffolds, providing chemists
with a general approach to the chemoselective oxidation of
equatorial alcohols for the first time.
Table 1. Oxidation with Isolated N−HVI
entry
N-HVI
solvent
T (°C)
yield (%)
a
1
2
3
4
5
6
7
8
9
Py-HVI (5)
Py-HVI (5)
Py-HVI (5)
Py-HVI (5)
Py-HVI (5)
Py-HVI (5)
2-MePy (8)
4-OMe (9)
4-NMe2 (10)
4-CF3 (11)
DCE
DCE
DCE
DCE
ACN
THF
DCE
DCE
DCE
DCE
r.t.
60
60
60
60
60
60
60
60
60
37
96
84 (95)
52 (84)
b
d
c
e
97
45
97
89
75
a
a
a
a
10
63
a
b
NMR yield with CH2Br2 as an internal standard 1.5 equiv of Py-
c
d
e
HVI. Yield after 8 h. 1.0 equiv of Py-HVI. Yield after 12 h.
in variable yields of 7 along with significant amounts of silyl-
protected 6. The use of the bulkier TBSOTf as the silyl
activator suppressed alcohol protection, and a screen of
hydrogen bonding and Brønsted acid additives8,9,16−18 found
that the use of AcOH gave a consistently excellent yield of 7
Scheme 2. Oxidation with in-Situ-Generated Py-HVI
To begin our study, efficient conditions for direct alcohol
oxidation with N-HVIs needed to be established. Using 4-
phenyl-2-butanol (6) as a model substrate, treatment with 2.0
equiv of Py-HVI in 1,2-dichloroethane (DCE) at room
temperature gave a modest 37% yield of the corresponding
ketone (7) (Table 1, entry 1). Increasing the temperature to
60 °C resulted in near-quantitative oxidation in just 2.5 h,
producing 7 in 96% isolated yield (entry 2). Decreasing the
equivalents of Py-HVI to either 1.0 or 1.5 (entries 3 and 4)
also gave high conversion but with decreased efficiency, taking
8 and 12 h to reach completion, respectively, and thus 2.0
equiv of Py-HVI was used for the remainder of our studies. A
brief solvent screen found that acetonitrile was equally effective
(entry 5), obviating the need for a halogenated solvent if so
desired. To gain more insight into the role of the nitrogen
ligand, a small library of electronically and sterically diverse N-
HVIs was screened. The use of ortho-substituted (8) or more
electron-rich (9, 10) N-HVIs also gave excellent yields (entries
7−9), whereas the highly reactive p-CF3-Py-HVI (11) was not
as effective (entry 10).
We then wished to further simplify our method by
developing a one-pot protocol via the in situ generation of
Py-HVI, avoiding the need for its isolation and storage. This
would also benefit the broader application of N-HVIs because
these reagents can be moisture-sensitive and prone to
degradation upon prolonged storage.9 The initial application
of our standard conditions for N-HVI synthesis using
TMSOTf, followed by the addition of the substrate, resulted
optimization details.) Several control reactions were conducted
to determine if Py-HVI was, in fact, the active oxidant under
these in situ conditions (Scheme 2, inset). Neither PhI(OAc)2
nor PhI(O2CCF3)2 alone produced any oxidation product, and
PhI(OAc)2/AcOH gave only trace oxidation, indicating that
Brønsted acid activation alone was insufficient. Finally, the
exclusion of pyridine from the reaction resulted in significant
substrate decomposition, likely through the reaction of highly
reactive [PhI(OAc)]OTf, formed upon the treatment of
PhI(OAc)2 with R3SiOTf.7b,17
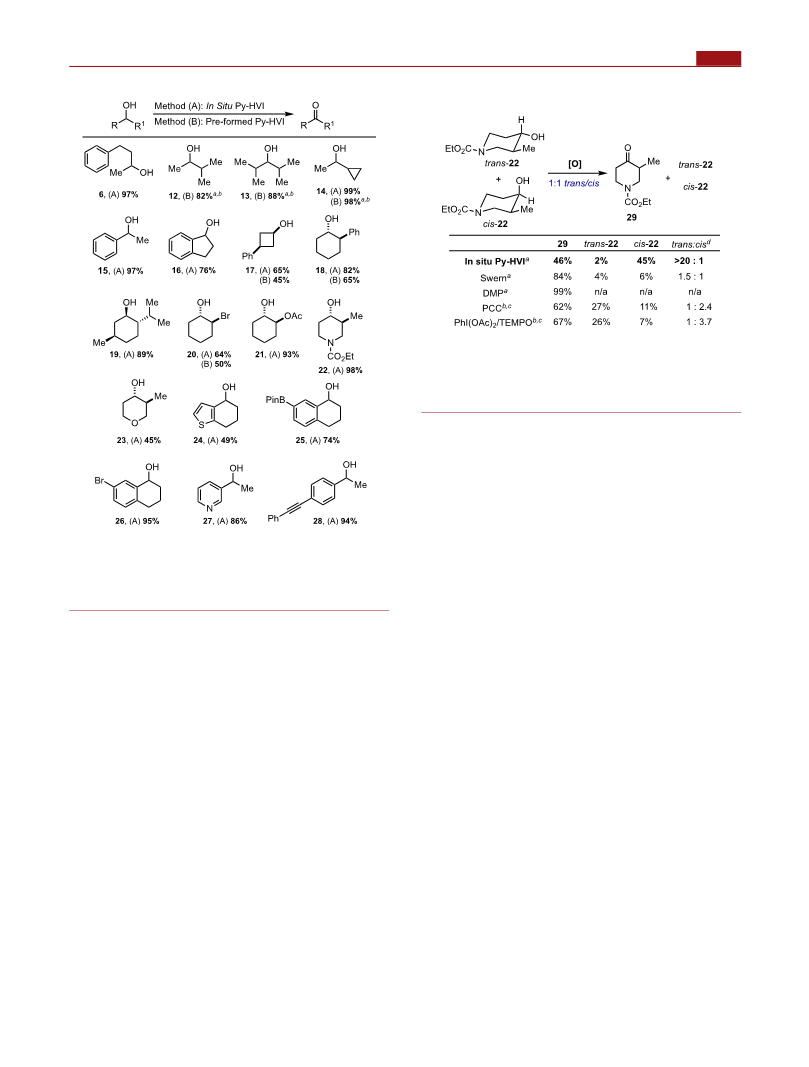
The scope of the oxidation was then examined using both in-
situ-generated (condition A) and isolated Py-HVI (5)
(condition B) and was found to be quite general (Scheme
3). A variety of acyclic, cyclic, and benzylic alcohols (6, 12−
19) gave good to excellent yields. The excellent yield of
cyclopropyl substrate 14 provides evidence that the reaction
does not proceed through a radical pathway. More function-
alized substrates, including those with halogens (20, 26),
acetate (21), alkyne (28), and saturated heterocycles (22, 23),
all proceeded in high yield. We were pleased to see that an
B
Org. Lett. XXXX, XXX, XXX−XXX

 Mikhael, Myriam
Mikhael, Myriam