5608
O. Cakmak et al. / Tetrahedron 58 (2002) 5603±5609
4.4.3. Entry 7. 1-BN 02.07 g, 10 mmol) was dissolved in
CCl4 040mL) in photochemical apparatus 060mL). To the
magnetically stirred solution was added bromine 03.98 g,
25 mmol) in CCl4 010mL) within 30min at 30 8C while
irradiating by projector lamp 0250W). The reaction
conversion, the solvent was removed by vacuo. The pre-
cipitate was ®ltered by a short silica gel 05 g) column by
1
eluting petroleum ether. H NMR analysis of the crude
material indicated existence of 1,5-DBN 4 and 1,3,5-TBN
5 in a ratio of 89:11, respectively.
1
progress was monitored by H NMR. After the completion
of reaction 0total 2 h) and the removing the solvent, the H
1
4.7.1. Preparation of 1,3,5-tribromonaphthalene 05).
Pentabromide 7 04.0g, 7.6 mmol) in dry THF 025 mL)
was treated with potassium tert-butoxide 04.154 g,
18.4 mmol, 2.5 equiv.) in dry THF 025 mL). The mixture
was stirred at room temperature overnight. After worked
up 03£50mL ether), dried and evaporated, the residue was
®ltered by using a short silica gel 010g) column eluting with
light petroleum ether. 1,3,5-Tribromonaphthalene 05) was
obtained and the product was recrystallized from chloro-
form±light petroleum in a yield of 91% 02.56 g) as a colour-
less solid, mp 107±1088C; 0Found: C, 32.72; H, 1.18.
C10H5Br3 requires C, 32.91; H, 1.35%); nmax 0KBr) 3073,
1606, 1479, 1353, 1189, 1079, 877, 819; dH 0400 MHz,
CDCl3) 8.42 01H, s, H4), 8.19 01H, d, J8.0Hz, H 8), 7.95
01H, s, J1.6 Hz, H2), 7.85 01H, d, J8.0Hz, H 6), 7.43 01H,
t, J8.0Hz, H 7); dC 0100 MHz, CDCl3) 134.06, 134.02,
132.26, 132.14, 129.99, 128.15, 127.73, 124.14, 122.20;
m/z 0CI, NH3) 367 095 MH1), 286 0100), 236 015), 206
025), 69 015%).
NMR analysis indicated formation of 1,5-DBN and the
pentabromide 7 in a ratio of 69:31, respectively.
4.4.4. Entry 8. To the magnetically stirred solution of 1-BN
01.33 g, 7.5 mmol) in CCl4 025 mL) in a photochemical
apparatus 045 cm3) at 08C 0ice bath) was added bromine
01.8 g, 11.25 mmol) in CCl4 015 mL) within 1 h while
irradiating by a projector lamp 0250W). The reaction
1
progress was monitored by H NMR. Reaction mixture
was totally irradiated for 2.5 h. After removing the solvent
and excess bromine, the residue was ®ltered on a short
Al2O3 column 010g) eluted with hexane. The 1H NMR
analysis of the residue indicated the formation of 1,5-
DBN and 1,3,5-TBN in a ratio of 92:8. Repeated crystal-
lization of the reaction mixture from CH2Cl2±petroleum
ether 01:2, 100 mL) in the freezer at 02158C) in 3 days
gave 1,5-DBN 01.47 g, 80%) in a pure form, 0pale yellow
crystals, mp 129±1308C 0lit.22 1318C); nmax 0KBr) 3060,
1795, 1488, 1322, 1189, 900, 701; dH 0250MHz, CDCl 3)
8.24 02H, d, J8.0Hz, ArH), 7.84 02H, d, Hz, ArH), 7.46
02H, t, J8.0ArH); dC 050MHz, CDCl 3) 135.07, 132.89,
129.39, 129.31, 125.02.
Same dehydrobromination procedure was repeated for
pentabromide 6. A product mixture was obtained, which
unable to isolate and to identify.
4.5. Bromination of naphthalene
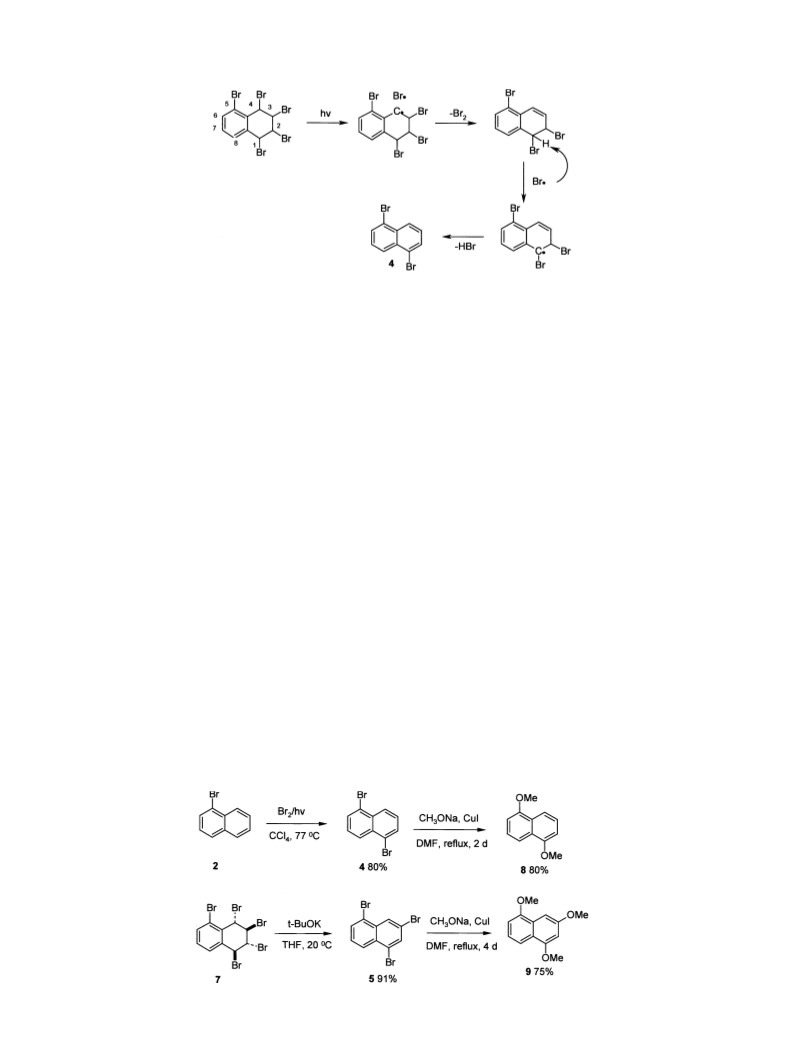
4.7.2. Preparation of 1,5-dimethoxynaphthalene 08).
Freshly cut sodium 04.84 g, 210.4 mmol) was added under
nitrogen gas to dry methanol 030mL). When dissolution
was complete, the warm solution was diluted with dry
dimethylformamide 0DMF, 60mL) followed by the addi-
tion of vacuum-dried cuprous iodide 0960mg, 5.0mmol).
After dissolution, 1,5-DBN 4 02.32 g, 8.32 mmol) in dry
DMF 030mL) was added. The reaction mixture was stirred
magnetically for 2 days under a nitrogen gas atmosphere at
re¯ux 0ca. 908C). After cooling to room temperature, H2O
0100 mL) and diethyl ether 0100 mL) were added to the
reaction mixture. The organic layer was separated, washed
with H2O 03£35 mL), and dried over CaCl2. The solvent
was removed and the product was passed through a short
column packed with silica gel 015 g) with eluting petroleum
ether. Recrystallization from a mixture of CH2Cl2 and
petroleum ether 0bp 40±508C) in the refrigerator 058C)
yielded 1,5-dimethoxynaphthalene 08) 01.8 g, 80%) as
colourless needles, mp 180±1818C 0lit.23 182±1848C);
0Found: C, 76.67; H, 6.60. C12H12O2 requires C, 76.57; H,
6.43%); dH 0200 MHz, CDCl3) 4.01 06H, s, OCH3), 6.86
02H, d, J8.0Hz, C-2,6), 7.40 02H, t, J8.0Hz, C-3,7),
7.86 02H, d, J8.0Hz, C-4,8); dC 0100 MHz, CDCl3)
57.54, 106.57, 116.22, 127.15, 128.65, 157.27; m/z 0CI,
NH3) 189 0100 MH1), 173 050), 145 010), 115 075), 102
025%).
Naphthalene was treated with one and two equiv. of
bromine in CH2Cl2 at 2308C in a separate reaction ¯ask.
Substrate±solvent molar ratio was taken in a ratio of 1:15.
Reaction of naphthalene with one equiv. bromine was
completed in 2 days to give 1-BN and 1,4-DBN 3 in ratio
of 90:5, respectively, besides unreacted naphthalene 5%
1
0conversion 95%), as assigned H NMR spectroscopy. The
reaction of naphthalene with 2 equiv. of bromine was
complete in 3 days. After removing the solvent, precipitated
residue was recrystallized from CH2Cl2±petroleum ether.
1,4-Dibromonaphthalene 03) was obtained in a yield of
90%. Reaction conditions and crystallization procedure
were the same as described in entry 3.
4.6. Heating pentabromides 6 and 7
A solution of a mixture of pentabromides 6 and 7 0120mg,
0.225 mol) in CCl4 07 mL) in a sealed glass-tube was heated
at 708C for 7 days in the dark. H NMR analysis of the
1
reaction mixture indicated that pentabromides 6 and 7
remained without change.
4.7. Irradiation of pentabromides 6 and 7
A mixture of pentabromide 6 and 7 0527 mg, 1 mmol) was
dissolved in CCl4 030mL) in a 50mL of reaction ¯ask. To a
magnetically stirred solution at re¯ux temperature of CCl4
was irradiated externally with projector lamp 0250W) for
2 h. The reaction progress was monitored by TLC or H
NMR. Evolving hydrogen bromide was observed and
bromine appeared during reaction. After completion of the
4.7.3. Preparation of 1,3,5-trimethoxynaphthalene 09).
Freshly cut sodium 00.74 g, 32 mmol) was added to dry
methanol 030mL) under nitrogen gas. After complete dis-
solution, dry dimethylformamide 0DMF, 30mL) was added,
followed by vacuum dried cuprous iodide 00.52 g,
2.7 mmol) and 1,3,5-TBN 5 01.0g, 2.7 mmol) in dry DMF
1

 Cakmak, Osman
Cakmak, Osman