M. Biesemans et al.
ꢀ
ꢀ
(P H)(1ꢀt)[{P (CH2)11SnBuCl}2O]t/2 (6): Elemental analysis calcd (%) for
t=0.26: H 7.92, C 71.11, Sn 15.35, Cl 4.59; found: H 7.90, C 70.82, Sn
The residual tin contents in the esters recovered from the
transesterifications catalysed with 1, 3 and 9 were measured
by ICP/MS. They were very low, the values ranging from 3
to 4 ppm. This amount corresponds to a tin content of about
0.5 mg of tin per mole of octyl acetate, the tin content with
a homogeneous catalyst left completely in the reaction prod-
ucts being 120 mg. This clearly shows a dramatic decrease
by a factor of approximately 200 in the residual amount of
tin found in the reaction product upon switching from a ho-
mogeneous catalyst to a grafted counterpart, unambiguously
demonstrating the benefits of grafting organotin catalysts. In
the latter case, residual tin in the reaction product is indeed
only induced by the loss from the beads of 0.4% of grafted
tin per run. The use of supported organotin catalysts is thus
a pertinent solution to reduce the amount of organotins in
esters obtained by catalytic transesterifications, without low-
ering to a dramatic extent the transesterification rate.
ꢀ
ꢀ
14.59, Cl 6.23; Raman: n˜ =597 (w, Sn Buasym), 517 (w, Sn Busym),
345 cmꢀ1 (m, Sn Cl); IR: n˜ =603 cm (m, Sn-O-Sn).
ꢀ1
ꢀ
ꢀ
ꢀ
(P H)(1ꢀt)[P (CH2)4SnBuO]t (7): An aqueous solution of sodium hy-
droxide (0.1 mL, 4m) was added to a suspension of 1 (400 mg) in dry
THF (20 mL) at 658C. After 6 h, polymer 7 was filtered off and washed
successively with a mixture THF/H2O (50/50), THF and then ethanol. El-
emental analysis (%) found: H 7.36, C 70.59, Sn 16.00, O 3.10, Cl 0.12;
see Table 1.
ꢀ
ꢀ
(P H)(1ꢀt)[P (CH2)11SnBuO]t (8): This compound was prepared by a
similar procedure to that for 7. Elemental analysis (%) found: H 8.46, C
71.59, Sn 11.35, O 2.76, Cl 3.41; see Table 1.
ꢀ
ꢀ
(P H)(1ꢀt)[P (CH2)4SnCl3]t (9): Diisopropylamine (1.39 g, 13.6 mmol)
and nBuLi (13.6 mmol) were successively added to dry THF (10 mL) at
08C. After 15 min, Cy3SnH (5 g, 13.6 mmol) was added slowly and the
mixture was stirred for 30 min. This solution of Cy3SnLi was slowly
ꢀ
ꢀ
added to (P H)(1ꢀt)[P (CH2)4Cl]t (3 g) suspended in dry THF (20 mL).
The mixture was stirred for 15 h at room temperature. After filtration,
ꢀ
ꢀ
(P H)(1ꢀt)[P (CH2)4SnCy3]t was washed with THF/H2O (50/50; 40 mL),
THF (6ꢃ40 mL) and ethanol (2ꢃ20 mL). Elemental analysis calcd (%)
for t=0.23: H 8.60, C 77.85, Sn 13.55; found: H 8.65, C 77.57, Sn 13.11.
A solution of tin tetrachloride (0.57 g, 2.2 mmol) in dry toluene (10 mL)
Conclusion
ꢀ
ꢀ
was slowly added to a suspension of (P H)(1ꢀt)[P (CH2)4SnCy3] (2 g) in
dry toluene (10 mL) at room temperature. After 48 h in the dark, the
polymer was filtered and washed eight times with pentane and twice with
ethanol. Elemental analysis (%) found: H 5.97, C 58.86, Sn 14.85, Cl
16.15; see Table 2.
All catalysts screened are reasonably active in the reactions
under investigation. They are more active toward primary
rather than secondary or bulky primary alcohols and not re-
active toward tertiary alcohols or phenols, indicating that
steric factors play an important role in the reaction mecha-
nism. The length of the spacer also plays a significant role;
whereas no significant difference in activity was found be-
tween spacer lengths 4 and 6,[17] elongation of the spacer to
11 methylene groups has a beneficial effect whatever the
nature of the substituents on the tin atom. Among all cata-
lysts tested, trichlorotin-substituted target polymers led to
the best yields of ester, in the same range as the more effi-
cient soluble ones. They could be recycled at least seven
times without loss of activity. The residual amount of organ-
otin in the prepared esters, expressed in tin mass, was as low
as 3 ppm.
Catalysis experiments on transesterification reactions of ethyl acetate:
Ethyl acetate, used both as reactant ester and solvent, and the appropri-
ate alcohol were engaged in a molar ratio 7/1. The mixture of ethyl ace-
tate, the alcohol and the insoluble Amberlite-supported catalyst was re-
fluxed for 8, 24 or 48 h. The catalyst was filtered off and washed with
CHCl3, THF and ethanol. Ethyl acetate was distilled off from the reac-
tion mixture. The ratio initial alcohol/obtained ester was determined by
integration (ꢁ1%) of the respective CH2O 1H resonances or by GC.
IR and Raman spectroscopy: IR spectra were recorded on a Bruker
Equinox 55 FT-IR spectrometer, equipped with an MIR source, KBr
beam splitter and a DGTS detector, from dry KBr pellets (200 mg) with
about 5 mg of substance. The Raman spectra were recorded on a Perkin–
Elmer 2000 NIR FT-Raman spectrometer by using a Raman_dpy2 beam
with 310 mW power.
NMR spectroscopy: Samples used for the determination of the ratio ini-
tial alcohol/obtained ester were prepared by dissolving about 10 mg of
mixture in CDCl3 (0.5 mL). Quantitative 1H spectra were recorded on a
Bruker AMX500 instrument. The 119Sn HR-MAS spectra were recorded
on the same instrument (186.50 MHz) with a specially dedicated Bruker
1H/13C/119Sn HR-MAS probe equipped with gradient coils, by using full
rotors containing approximately 20 mg of resin beads, swollen in approxi-
mately 100 ml of CDCl3 and magic angle spinning at 4000 Hz. (CH3)4Sn
was used as internal reference. CP-MAS spectra were recorded on a
Bruker Avance 250 spectrometer, equipped with a 4 or 7 mm MAS
broad-band probe, operating at 89.15 MHz for 117Sn. The magic angle
was set by using a KBr sample. The chemical shift reference for the 117Sn
nucleus was set with (cyclo-C6H11)4Sn (ꢀ97.35 ppm relative to (CH3)4Sn).
The 117Sn CP-MAS spectra were acquired with 4 K data points over a
spectral width of 107 kHz, a relaxation delay of 2 s and 10000 to 40000
scans.
Experimental Section
Synthesis: The syntheses of 1, 2, 4 and 5 have already been reported.[17–19]
The syntheses of 3 and 6 were performed according to the same proce-
dure with Br(CH2)11Cl being used in the first reaction step instead of
Br(CH2)6Cl. Br(CH2)11Cl was prepared from Br(CH2)11OH (Aldrich) by
reaction with SOCl2, following an established literature procedure.[21] The
ensuing reaction steps were completely analogous to those used for 4.
ꢀ
ꢀ
(P H)(1ꢀt)[P (CH2)11Cl]t: Elemental analysis calcd (%) for t=0.29: H
8.96, C 84.47, Cl 6.57; found: H 8.93, C 84.34, Cl 5.97; IR: n˜ =651 cmꢀ1
1
ꢀ
(w; CCl); H HR-MAS NMR: d=3.49 ppm ( CH2Cl)
ꢀ
ꢀ
(P H)(1ꢀt)[P (CH2)11SnBuPh2]t: Elemental analysis calcd (%) for t=
0.26: H 8.07, C 78.48, Sn 13.45; found: H 7.87, C 78.27, Sn 12.61, Cl
ꢀ
ꢀ
ꢀ
<0.2; IR: n˜ =1074 (m, Sn Ph), 727 (w, C Cmono), 656 (w, Sn Crock), 594
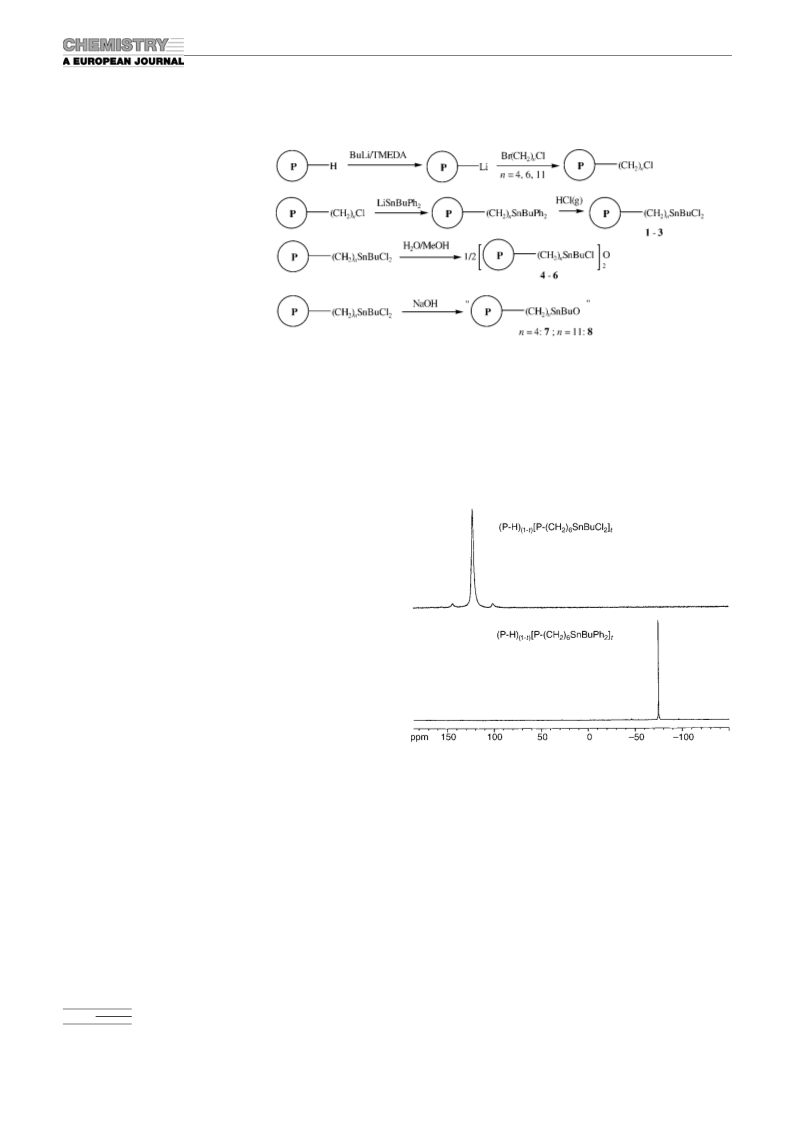
(w, Sn Buasym), 510 cmꢀ1 (w, Sn Busym); 119Sn HR-MAS NMR: d=
ꢀ71 ppm.
ꢀ
ꢀ
Acknowledgements
ꢀ
ꢀ
(P H)(1ꢀt)[P (CH2)11SnBuCl2]t (3): Elemental analysis calcd (%) for t=
The financial support (Scholarship) of Consejo Nacional de Ciencia y
Tecnologia (CONACYT-Mexico) and Universidad Autonoma Metropoli-
tana-X are gratefully acknowledged (C.C.-C.). M.B. and R.W. are indebt-
ed to the Fund for Scientific Research, Flanders (Belgium; FWO, Grant
0.26: H 7.65, C 68.64, Sn 14.84, Cl 8.87; found: H 7.71, C 68.49, Sn 13.77,
Cl 8.72; Raman: n˜ =596 (w, Sn Buasym), 520 (w, Sn Busym), 347 cmꢀ1 (m,
ꢀ
ꢀ
Sn Cl); 119Sn HR-MAS NMR: d=126 ppm.
ꢀ
2460
ꢂ 2005 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Chem. Eur. J. 2005, 11, 2455 – 2461

 Camacho-Camacho, Carlos
Camacho-Camacho, Carlos