1178
G. Catucci et al. / Biochimica et Biophysica Acta 1864 (2016) 1177–1187
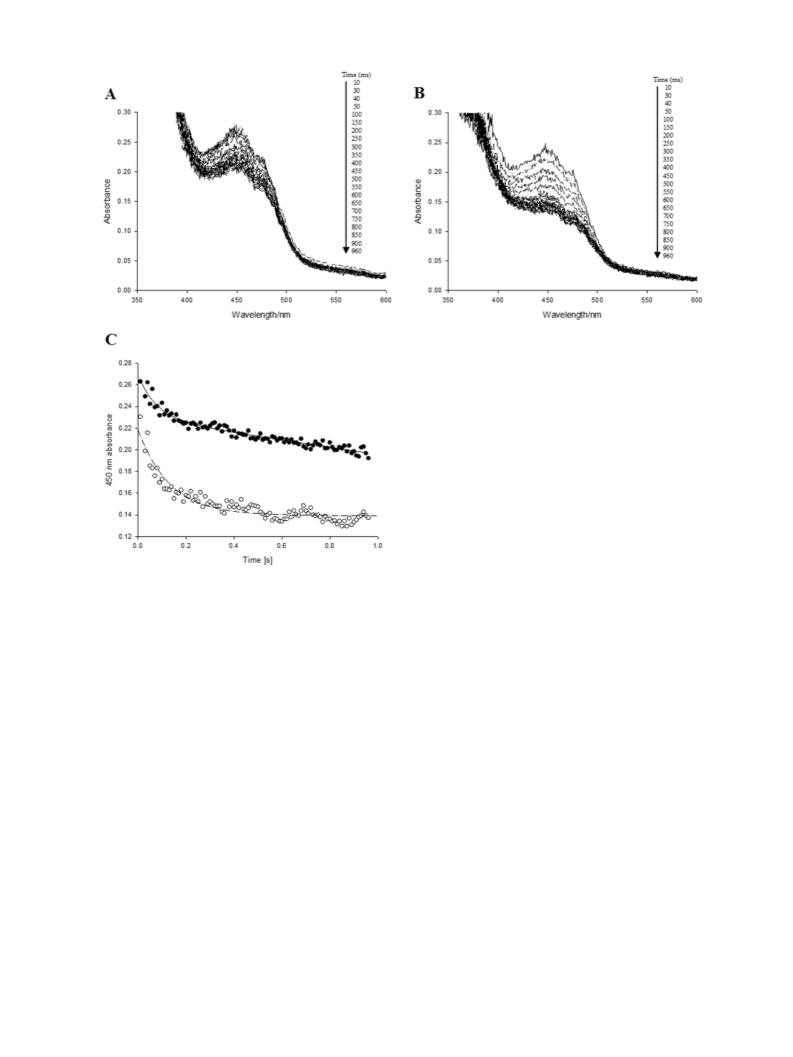
atom of molecular oxygen being transferred to the substrate with the
other forming water. The formation of the reactive peroxyflavin is inde-
pendent of substrate binding, distinguishing BVMO enzymes from other
flavin-containing hydroxylases and cytochromes P450. In the reaction,
the nucleophilic attack of the flavin peroxide on the carbonyl group of
the substrate forms a tetrahedral species that is known as a Criegee in-
termediate, and this rearranges to form an ester that is stereochemically
imposed by the enzyme. The product then leaves the active site
followed by the release of the NAD(P)+ and the cycle can start over
again [4–6].
ethyl acetate, methanol, n-hexane, and salts were purchased from
Sigma–Aldrich (Italy).
2.2. Cloning of Ar-BVMO gene in pT7 expression vector
A. radioresistens S13 was isolated from soil surrounding an activated
sludge pilot plant (Torino, Italy) and its Ar-BVMO gene was previously
isolated by our group from A. radioresistens S13 genome by PCR using
two primers designed on the basis of the almA gene of this species
[13]. The product of this PCR was used as the template for the insertion
of the desired restriction sites upstream and downstream of the gene for
cloning into pT7-7 [17,18].
The BVMO gene was amplified with a nested PCR using primers spe-
cifically designed on the basis of the Ar-BVMO coding gene, adding a 6
histidine tag at the C-terminus for ease of purification. The sequences
of the primers used were: 5′-AACCAACCAAGGATCCATGGATAAACAC
ATTGATGTTCTAATT-3′ and 5′-TTGGTTGGTTGAATTCTTAATGGTGATG
GTGATGGTGTGAAACCAGTTTAGGTTTACGTTC-3′. The PCR conditions
were as follows: denaturation at 94 °C for 3 min; 30 cycles at 94 °C for
30 s, 64 °C for 30 s, and 72 °C for 1 1/2 min; and a final extension at
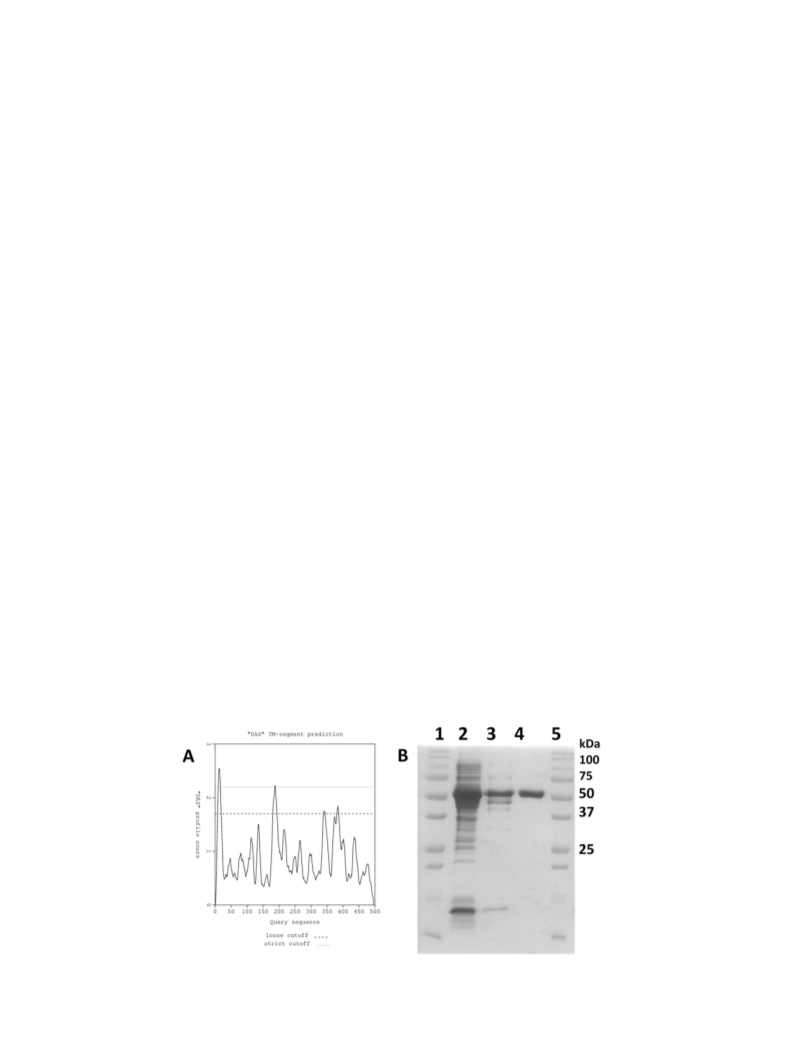
72 °C for 10 min. A PCR product of the expected size (1541 bp) was ob-
tained. The PCR product was digested with EcoRI and BamHI restriction
enzymes. The linearized insert and pT7-7 vector [17,18], with compati-
ble sticky ends, were used in a ligation reaction to produce the
desired clone. Escherichia coli BL21 (DE3) competent cells were then
transformed with the ligation mixtures and plated on LB agar with
100 μg/mL ampicillin. Colonies with the insert were screened and sub-
sequently the entire pT7-BVMO sequenced.
The first BVMO to be studied in detail was cyclohexanone
monooxygenases (CHMO) from Acinetobacter sp. (NCIMB 9871) [7,8].
Although the genus Acinetobacter has been shown to be a source of sev-
eral BVMO proteins, many of these enzymes are not available in recom-
binant form and therefore have not yet been characterized. Using
sequence homologies and the presence of the conserved Baeyer-
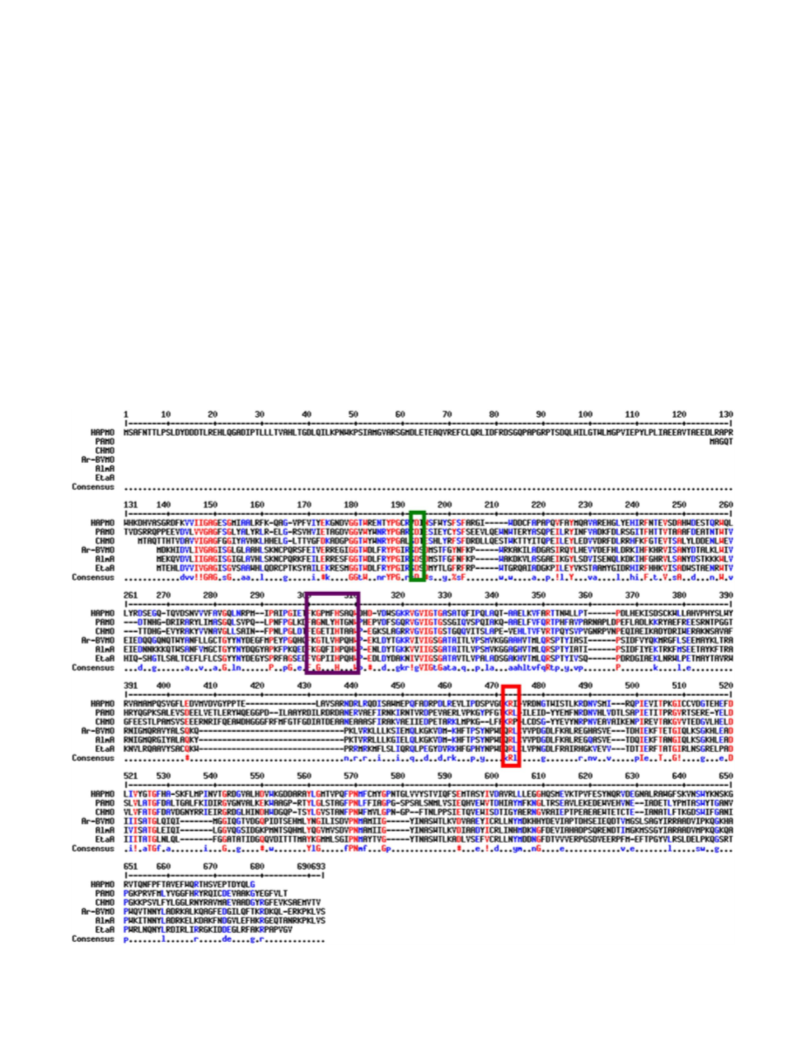
Villiger sequence motif, we have recently discovered a novel BVMO,
Ar-BVMO, in the genome of Acinetobacter radioresistens S13 [9]. This
protein was found to share a 49.7% sequence identity and 71.0% se-
quence similarity with the ethionamide-activating (EtaA) BVMO from
Mycobacterium tuberculosis [10–12]. Furthermore, phylogenetic analysis
of the Ar-BVMO not only placed this protein in the same branch as EtaA
but also showed it to be closely related to the phase-1 drug metabolizing
enzyme, human flavin-containing monooxygenase 3 (FMO3), but only
distantly related to other canonical bacterial BVMO proteins [9,13].
Considering these relationships, initially we studied Ar-BVMO for its
possible activities with “unusual/non-typical” substrates and demon-
strated that it is not only capable of oxidizing two anticancer drugs
metabolized by human FMO3, Danusertib and Tozasertib [14,15], but
it can also oxidize other synthetic drugs such as imipenem [2].
Imipenem is a carbapenem, which is a clinically important antibiotic
family used in the treatment of MultiDrug-Resistant (MDR) bacterial in-
fections. Interestingly, an identical gene to that of Ar-BVMO also exists
in Acinetobacter Baumannii, one of the so-called superbugs [2]. In these
cases, Ar-BVMO seems to be responsible for the de-activation of this an-
tibiotic, and therefore it acts more like a drug metabolizing enzyme.
Chen and coworkers [16] have also shown that a FMO from Methylocella
silvestris is able to oxidize trimethylamine, which is also a typical sub-
strate of eukaryotic FMO enzymes. The reason(s) why prokaryotic flavin
monooxygenases should metabolize substrates that are typically
metabolized by the eukaryotic FMO enzymes but not present in
prokaryotic natural habitats is still not clear.
2.3. Expression of wild-type Ar-BVMO
The expression of the Ar-BVMO enzyme was carried out in a 5 L
Biostat A plus fermenter. Forty milliliters of overnight liquid culture of
E. coli BL21 (DE3) cells transformed with pT7-BVMO were used to inoc-
ulate 4 L LB medium supplemented with ampicillin (100 μg/mL) and ri-
boflavin (20 mg/L). The cells were grown at 37 °C and 200 rpm until
OD600 of 0.5 was reached. At this point, protein expression was induced
by the addition of IPTG and the temperature was lowered to 24 °C. The
cells were harvested 20 h post-induction by 20 min centrifugation at
4000 rpm at 4 °C.
In order to better understand these observations, we have undertak-
en the characterization of the wild type and mutant recombinant Ar-
BVMOs. Kinetic studies of Ar-BVMO, as well as investigations into the
roles of its active site amino acids thought to be important for BV reac-
tions were carried out. Comparisons of these properties with those of
canonical BVMO enzymes were used to try to understand how Ar-
BVMO is competent to carry out oxygenations with synthetic and
medically relevant drugs.
The role of the active site arginine 292 was also investigated in
relation to the enzyme activity towards linear ketones. Two mutants
R292A and R292G have no BV activity while still maintaining their S-
and N-oxygenation capabilities. These monooxygenation activities are
important for human drug metabolism, and therefore, these Ar-BVMO
mutants could be useful as bacterial biocatalysts for large-scale produc-
tion of human drug metabolites.
2.4. Purification of wild-type Ar-BVMO
All purification procedures were carried out at 4 °C. The pellet was
resuspended in 50 mL of Buffer A (50 mM KPi pH 7.4, 20% glycerol,
5 mM β-mercaptoethanol) supplemented with 0.5 mM of the protease
inhibitor phenylmethylsulfonyl fluoride (PMSF) and 1 mg/mL of lyso-
zyme. The resuspended cells were disrupted by sonication. The lysate
was supplemented with 1% Igepal (octylphenoxypolyethoxyethanol),
stirred for 30 min at 4 °C to obtain complete homogeneity, and then cen-
trifuged at 4000 rpm for 45 min. The clarified lysate was loaded onto a
DEAE-Sepharose Fast-Flow column pre-equilibrated with buffer A. The
bound BVMO protein was extensively washed with the same buffer be-
fore it was eluted by applying a gradient of 0–250 mM NaCl also in buff-
er A. Fractions containing the flavoprotein were then pooled and loaded
on a Nickel chelating Sepharose Fast-Flow column pre-equilibrated
with Buffer B (Buffer A supplemented with 250 mM NaCl). The bound
protein was washed with 5 column volumes of Buffer B, 5 column vol-
umes of Buffer B supplemented with 1 mM histidine and 2 column vol-
umes of Buffer B supplemented with 5 mM of histidine. The protein was
finally eluted with Buffer B supplemented with 40 mM histidine.
Fractions presenting the characteristic FAD peaks were pooled and
buffer exchanged with Amicon centrifugal units (30 kDa cut off) with
100 mM potassium phosphate buffer pH 7.4, 20% glycerol.
2. Materials and methods
2.1. Reagents
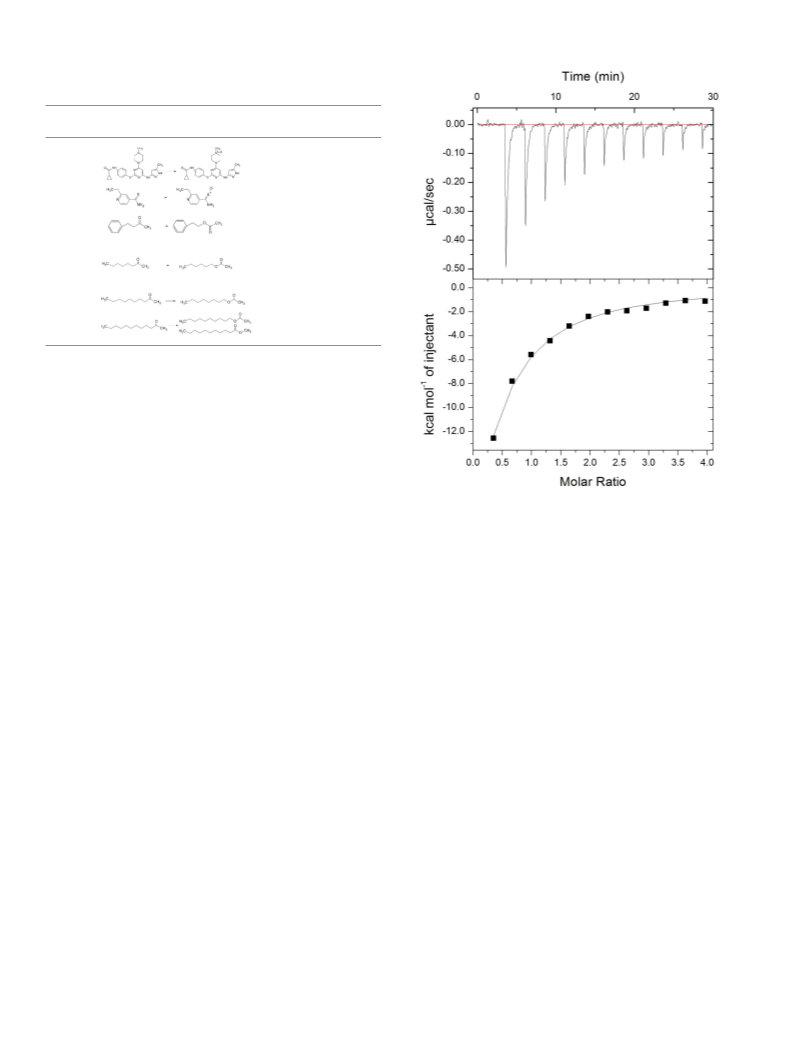
Riboflavin, NADPH, NADH, ethionamide, 4-phenyl-2-butanone,
phenethyl acetate, 2-octanone, hexyl acetate, 2-decanone, octyl acetate,
2-decanone, methyl undecanoate, decyl acetate, acetonitrile,

 Catucci, Gianluca
Catucci, Gianluca