4592
B. V. Subba Reddy et al. / Tetrahedron Letters 46 (2005) 4589–4593
bridged dimeric species 3 on the basis of much analogy9
and especially because the closely related sulfur analog
(Me2AlSMe)2 has been determined to be this type of
S-bridged dimer.10 On the basis of this structure a most
intriguing possibility emerges for the mechanism of the
mild and efficient cleavage of methyl esters by the new
reagent 3. This mechanistic pathway is summarized in
Scheme 1. In this pathway the methyl ester is activated
by coordination of the COOMe carbonyl oxygen to an
aluminum of 3 leading after cleavage of one of the Al–
Te bridge bonds to the putative intermediate 4. Cleavage
of the Me–O bond in 4 could then occur by an unusual
intramolecular backside displacement on methyl by the
distal TeMe subunit, as shown. Because of the number
of bonds in the path between the methyl group being
attacked and the terminal Te nucleophile and the length
of the bonds to Te, a stereoelectronically favorable
colinear O–Me–Te–Me transition state is available from
4.11 This would represent an extremely rare and singular
example of an intramolecular backside nucleophilic dis-
placement reaction in which the nucleophile is linked to
the leaving group. Of course, it is also possible that a
two-step decomposition pathway occurs from 4: termi-
nal MeTe–Al bond dissociation to form MeTeꢀ which
then attacks the methyl ester Me group as an external
nucleophile to produce the ester cleavage product.
Whichever of these two alternatives operates, it is clear
that the mechanistic pathway shown in Scheme 1 pro-
vides a simple explanation of the unique reactivity of
the reagent 3 with hindered carboxylic acid methyl es-
ters. It is also apparent that the process of methyl ester
cleavage outlined in Scheme 1 represents a new para-
digm for ester deprotection.
A similar explanation can be used for the cleavage of
aromatic methyl ethers by 3, except that in this case
the two-step pathway for Me–O bond cleavage from
the complex corresponding to 4 becomes much more
likely for stereoelectronic reasons. Finally, the deacetyl-
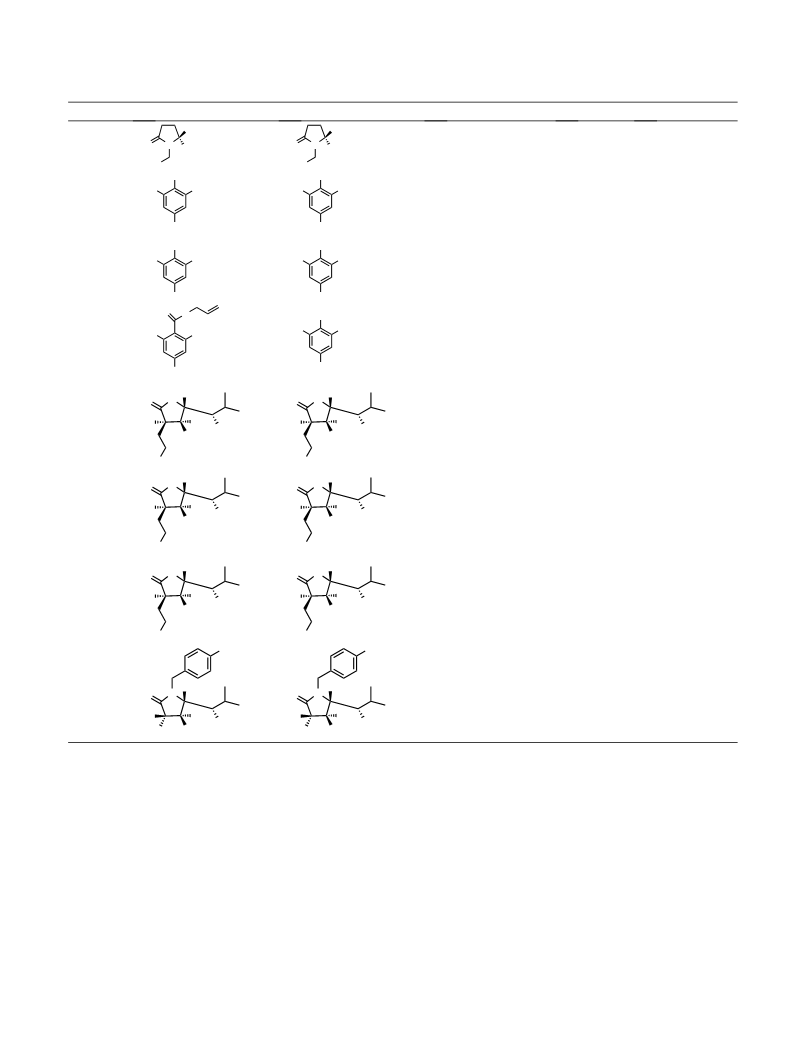
ation reactions induced by the reagent 3 (Table 2, entries
1–4) can be explained by attack on the acetate carbonyl
of complex 5 by the distal MeTe group.
Me Me
Me
Me
Al
O
Te
Al
O
Te
Me
Me
Te Me
O
R
Me
Me
RO AlMe2
+
5
H3O+
ROH
MeCOOH
+
References and notes
1. Feling, R. H.;Buchanan, G. O.;Mincer, T. J.;Kauffman,
C. A.;Jensen, P. R.;Fenical, W. Angew. Chem., Int. Ed.
2003, 42, 355–357.
2. Reddy, L. R.;Saravanan, P.;Corey, E. J. J. Am. Chem.
Soc. 2004, 126, 6230–6231.
3. Reddy, L. R.;Fournier, J.-F.;Reddy, B. V. S.;Corey, E. J.
J. Am. Chem. Soc., submitted for publication.
4. For example, none of the carboxylic acid corresponding to
2 could be obtained using sodium, lithium, barium, or
lanthanum hydroxides using a variety of solvents and
temperatures. Aqueous acids and Lewis acidic conditions
(e.g., BCl3, Me3LiI) also failed. For reviews on methyl
ester cleavage, see: (a) Greene, T. W.;Wuts, P. G. M.
Protective Groups in Organic Synthesis, 3rd ed.;John
Wiley and Sons: New York, 1999;(b) Kocienski, P. J.
Protecting Groups, 3rd ed.;George Thieme: Stuttgart,
New York, 2004;(c) Salomon, C. J.;Mata, E. G.;
Mascaretti, O. A. Tetrahedron 1993, 49, 3691–3734;(d)
Nicolaou, K. C.;Estrada, A. A.;Zak, M.;Lee, S. H.;
Safina, B. S. Angew. Chem., Int. Ed. 2005, 44, 1378–1382;
(e) Olah, G. A.;Narang, S. C.;Salem, G. F.;Gupta, B. G.
B. Synthesis 1981, 142–143;(f) Marchand, P. S. J. Chem.
Soc., Chem. Commun. 1971, 667–668;(g) Bartlett, P. A.;
Johnson, W. S. Tetrahedron Lett. 1970, 4459–4462.
5. For the analogous sulfur reagent, see: (a) Corey, E. J.;
Kozikowski, A. P. Tetrahedron Lett. 1975, 925–928;(b)
Corey, E. J.;Beames, D. J. J. Am. Chem. Soc. 1973, 95,
5829–5830;(c) Hatch, R. P.;Weinreb, S. M. J. Org. Chem.
1977, 42, 3960–3961.
Me
O
Me
Me
Me
Me
Te
R
C
+
Al
Al
O
Me
Te
Me
3
Me Me Me
Al Te
Al
Te
Me
O
Me
Me
R
C
O
Me
4
6. For the corresponding selenium reagent, Me2AlSeMe,
prepared by reaction of selenium with Me3Al, see:
Kozikowski, A. P.;Ames, A. J. Org. Chem. 1978, 43,
2735–2737.
7. At the end of the 6 h reaction time all the gray-white Te
powder had dissolved and only a small amount of an
insoluble colorless solid remained at the bottom of the
flask (possibly due to the presence of impurity in the Te
powder used). The clear supernatant solution of Me2Al-
TeMe was then drawn off by syringe as needed.
Me
Me
O
Al
R
C
Te Me
Me
MeTeMe
+
O
Al
Me
H3O+
RCOOH
8. A solution of methyl mesitoate (1.78 g, 10 mmol) in a
2 mL of toluene was added to a stirred solution of freshly
prepared dimethylaluminum methyltellurolate (3, 15 mL;
12 mmol;0.8 M solution) in toluene. The mixture was
Scheme 1. Possible mechanistic pathway for the facile Me–O bond
cleavage of methyl esters by 3.

 Reddy, B.V. Subba
Reddy, B.V. Subba