Chemistry Letters 2002
1021
Table 2. Conversion and product distribution in liquid-phase hydrodechlorination of CCl4
Conversion
of CCl4
Product distribution (mole %)
Conversion
of C2H5OH
Product distribution (mole %)
EVE
Run #
Catalysts
CHCl3
C2HxCla4ꢁx
1.3
1.2
C2Cl6
DEE
AA
DEC
1
2
Pd/Mont
Pd/AC
2O3
43.5
47.8
93.3
97.2
5.4
1.6
17.3
9.9
77.4
30.7
12.1
36.7
8.8
32.2
1.7
0.4
3Pd/Al
4
40.391.0
32.9
22.8
2.9
6.1
12.2 .7
4349.0
7.1
0.2
Pd/SiO2-Al2O3
Pd metal
PdCl2
90.4
72.8
90.2
95.6
98.9
4.7
5.1
4.7
0.9
0.6
4.9
22.1
5.1
17.6
7.4
68.4
55.4
36.7
71.0
61.6
25.8
25.5
56.3
7.6
5.3
8.6
0.5
10.5
3.3
5
6
24.1
48.4
9.4
3.7
7
Pt/Mont
Pt/AC
3.5
0.5
19.7
5.2
18.9
22.4
2.5
3.7
8
30.5
12.3
9
10
Pt/Al2O3
Pd/Mont
Pd/AC
12.375.5
36.7
55.0
5.3 19.2
5.344.0
51.2.9
3 0.9
91.9
96.1
95.3
2.5
1.9
1.5
5.6
2.0
3.2
20.0
.1 14.35319.7
18.5
67.4
17.8
68.8
14.0
12.4
7.4
8.1
11.2
10.7
11
12
9.4
Pt/Mont
31.4
13Pt/AC
24.395.3 2.7
2.0
11.2
57.0
1182.3.1
12.6
Reaction condition of Run 1 to 9: T ¼ 323 K; P(H2) ¼ 3:0 MPa; CCl4 ¼ 64:5 mmol; C2H5OH ¼ 217:4 mmol; internal standard (n-undecane) ¼ 1:3 mmol;
catalyst ¼ 0:1 g; reaction for 12 h. Reaction condition of Run 10 to 13: Same as Run 1 except for the P(H2) ¼ 1:5 MPa and P(O2) ¼ 0:1 MPa. Run 5 and 6:
Amount of palladium loading was equivalent to the surpported 5.0 wt% palladiumcatalyst of 0.1 g. aC2Cl4 and C2HCl3 were the main products in C2HxCl4ꢁx
(x ¼ 0, 1, 2). Abbreviation: DEE=1,1-diethoxyethane; AA=acetaldehyde; EVE=ethyl vinyl ether; DEC=diethyl carbonate.
isotope experiment with C2H5OD and CCl4. A large amount of
DCl with HCl was produced, but chloroform in the form of CDCl3
was not detected. Thus, C2H5OH is not directly involved in the
formation of CHCl3, but could exert the positive effects in activity
and selectivity by rapidly scavenging chlorine. This would also
increase the catalyst life because catalyst deactivation in HDC of
CCl4 is generally induced from the strongly adsorbed chlorine or
carbonaceous species.3{5
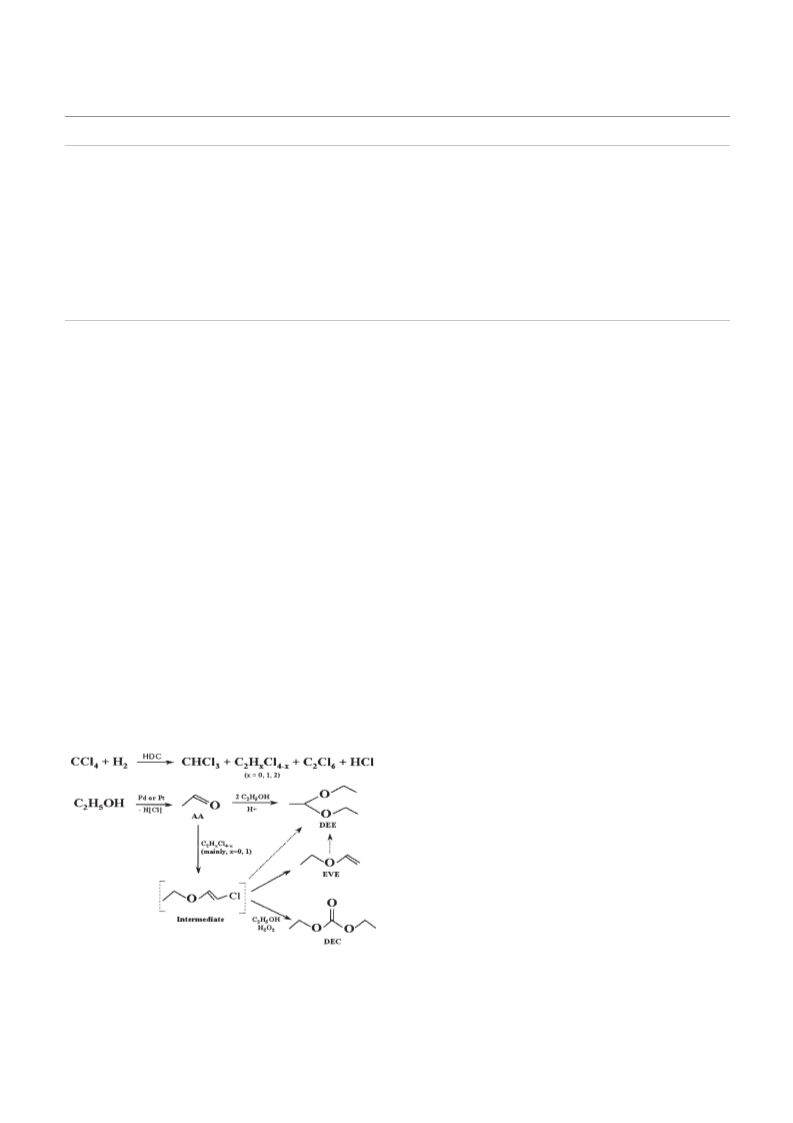
A possible reaction pathway to form DEC, EVE and DEE
during the hydrodechlorination of CCl4 in the presence of
C2H5OH is shown in Figure 1. Catalytic hydrodechlorination of
CCl4 proceeds according to the known pathway over Pt or Pd to
produce mainly CHCl3 with gaseous hydrogen as the hydrogen
source. As a protic solvent, C2H5OH could donate the proton to
chlorine produced in the main reaction, and turned itself to
acetaldehyde (AA) on Pd or Pt sites. The reaction of acetaldehyde
with ethanol produces DEE by acid-catalyzed aldol condensation.
Ethyl vinyl ether (EVE) could be formed from the reaction of AA
with C2HxCl4ꢁx (mainly, x ¼ 0 or 1). Furthermore, chlorine-
containing C=C group in EVE precursors could be easily
dechlorinated to DEE by hydrogen mainly from the proton in
C2H5OH adsorbed on the catalyst surfaces. Diethyl carbonate is
produced by the addition of oxygen to carbon-carbon double
bonds11 in an intermediate of chlorine-containing C=C group in
EVE precursors. The oxidizing agent may be hydrogen peroxide
generated in-situ on the transition metal by the reaction of
dihydrogen and dioxygen.12;13 When hydrogen peroxide was
added ex-situ at the beginning of reaction, the formation of DEC
was greatly enhanced. The oxidative cleavage of EVE inter-
mediates to produce DEC is only possible in acidic media, but no
reaction occurred only with C2H5OH itself even on the acidic
support. To enhance the generation of DEC in our phosgene-free
reaction conditions, the amount of reaction intermediates,
chlorine-containing EVE precursors with the terminal vinyl
group, should be an important variable. The larger are the
amounts of C2HxCl4ꢁx (mainly, x ¼ 0 or 1) and in-situ generated
hydrogen peroxide, the higher selectivity to DEC is expected.
In summary, catalytic hydrodechlorination of CCl4 over
supported Pd or Pt in the presence of ethanol gives not only the
selective synthesis of CHCl3, but also co-production of valuable
diethyl carbonate and 1,1-diethoxyethane.
We are grateful for the financial support provided by the
Brain Korea 21 project of Ministry of Education in 2002, Korea.
References
1
S. Y. Kim, H. C. Choi, O. B. Yang, K. H. Lee, J. S. Lee, and Y. G. Kim, J.
Chem. Soc., Chem. Commun., 1995, 2169.
2
H. C. Choi, S. H. Choi, J. S. Lee, K. H. Lee, andY. G. Kim, J. Catal., 166,
284 (1997).
3J. W. Bae, E. D. Park, J. S. Lee, K. H. Lee, Y. G. Kim, S. H. Yeon, and B.
H. Sung, Appl. Catal., A, 217, 79 (2001).
4
5
Z. C. Zhang and B. C. Beard, Appl. Catal., A, 174, 33 (1998).
C. D. Thompson, R. M. Rioux, N. Chen, and F. H. Ribeiro, J. Phys.
Chem. B, 104, 3067 (2000).
6
7
L. M. Gomez-Sainero, A. Cortes, X. L. Seoane, and A. Arcoya, Ind. Eng.
Chem. Res., 39, 2849 (2000).
M. A. Aramendia, V. Borau, I. M. Garcia, C. Jimenez, F. Lafont, A.
Marinas, J. M. Marinas, and F. J. Urbano, J. Catal., 187, 392 (1999).
M. A. Pacheco and C. L. Marshall, Energy Fuels, 11(1), 2 (1997).
J. X. Zhen, S. Y. Hua, and C. S. Hua, Catal. Lett., 69, 153(2000).
8
9
10 M. R. Capeletti, L. Balzano, G. de la Puente, and M. Laborde, Appl.
Catal., A, 198, L1 (2000).
11 F. C. Carey and R. J. Sundberg, ‘‘Advanced Organic Chemistry; Part
B,’’ 3rd ed., Plenum Press, New York (1993), p 624.
12 E. D. Park and J. S. Lee, Catal. Commun., 2, 187 (2001).
13C. Y. Shen, E. A. Garcia-Zayas, and A. Sen, J. Am. Chem. Soc., 122,
4029 (2000).
Figure 1. A proposed reaction pathway.

 Bae, Jong Wook
Bae, Jong Wook