G. Cavinato et al. / Journal of Molecular Catalysis A: Chemical 352 (2012) 63–69
69
depressuring to 1 atm the suspension was collected on a filter (IR
ꢀCO at 1822 for [Pd3(CO)3(PPh3)4] [35]. The GC analysis of the
solution showed the presence of DMC and of DMO in the ratio
ca. 15/1 and of trace amounts of DMS. Thus DMC may form also
from a Pd(COOMe)2 species via attack of MeOH to a Pd–COOMe
bond or by decarbonylation to a Pd(COOMe)(OMe) species, fol-
lowed by reductive elimination of DMC. Instead, it is unlikely that
DMS is formed through simultaneous insertion of ethene into two
Pd–COOMe bonds of a Pd(COOMe)2 species or insertion of ethene
into a Pd–COOMe moiety of a Pd(COOMe)2 species, with formation
of a Pd–(CH2CH2COOMe)(COOMe) intermediate, followed by the
transfer of the other COOMe moiety and elimination of DMS.
It is more reasonable that DMS is formed through the first steps
of the well known mechanism leading to PKs [2,4], i.e. the inser-
with a -chelate isomer), followed by the insertion of CO with the
formation of a Pd–COCH2CH2COOMe intermediate (which might
be in equilibrium with its ␥-chelate) [36], followed by the attack of
coordinated MeOH [37] to yield DMS.
initiators and directs the catalysis toward Pd–COOMe ones. In the
absence of BQ, DMS is not formed and catalysis takes a Pd–H route
to MP. The catalytic cycles for the formation of DMC, DMO, MP and
DMS are proposed on the bases of the results of the catalysis and of
model reactions in an NMR tube.
Acknowledgements
The financial support of MIUR (Rome) is gratefully acknowl-
edged. The authors thank Dr. Andrea Boareto of the Department
of Chemical Sciences of the University of Padua for mass spectra.
References
[1] A. Sen, Acc. Chem. Res. 26 (1933) 303.
[2] E. Drent, P.H.M. Budzelaar, Chem. Rev. 96 (1996) 663.
[3] M. Beller, A.M. Tafesh, in: B. Cornils, W.A. Hermann (Eds.), Applied Homoge-
neous Catalysis with Organometallic Compounds, vol. 1, VCH, Weinheim, 1996,
p. 187.
[4] C. Bianchini, A. Meli, Coord. Chem. Rev. 225 (2002) 35.
[5] G.P. Belov, E.V. Novikova, Russ. Chem. Rev. 73 (2004) 267.
[6] M. Beller (Ed.), Catalytic Carbonylation Reactions, Topics in Organometallic
Chemistry, vol. 18, Springer-Verlag, Berlin Heidelberg, 2006.
[7] E.G. Chepaikin, A.P. Bezruchenko, A. Ben’ei, F. Io, Izv. Akad. Nauk SSSR, Ser. Khim.
(1989) 743.
3.4.4. On the formation of DMO
DMO is probably formed from a di-carbomethoxy species.
As reaction model we studied the reactivity of trans-
[Pd(COOMe)2(PPh3)2] with BQ dissolved in CD2Cl2 in an NMR
tube, under 6 atm of CO and ethene (1/1). The complex starts
to react at 20 ◦C with formation of [Pd(BQ)(PPh3)2] and DMO.
Catalysis to DMO was observed at 60 ◦C. In the absence of BQ the
complex is stable at a temperature of 35 ◦C. It can be concluded that
(i) BQ destabilizes the starting complex, probably by promoting its
isomerization to one having the two carbomethoxy ligands in a
vicinal position, thus favouring the reductive elimination of DMO
and that (ii) under these conditions the elimination of DMO is
faster than catalysis.
[8] P. Kalck, M. Urrutigoity, O. Dechy-Cabaret, in: M. Beller (Ed.), Catalytic Carbony-
lation Reactions, Topics in Organometallic Chemistry, vol. 18, Springer-Verlag,
Berlin Heidelberg, 2006, pp. 97–123.
[9] G. Cavinato, L. Toniolo, A. Vavasori, in: M. Beller (Ed.), Catalytic Carbonylation
Reactions, Topics in Organometallic Chemistry, vol. 18, Springer-Verlag, Berlin
Heidelberg, 2006, pp. 125–164.
[10] W. Clegg, G.R. Eastham, M.R.J. Elsegood, R.P. Tooze, X.L. Wang, K.W. Whiston, J.
Chem. Soc. Chem. Commun. (1999) 1877.
[11] G.R. Eastham, B.T. Heaton, J.A. Iggo, R.P. Tooze, R. Whyman, S. Zacchini, J. Chem.
Soc. Chem. Commun. (2000) 609.
[12] W. Clegg, G.R. Eastham, M.R.J. Elsegood, B.T. Heaton, J.A. Iggo, R.P. Tooze, R.
Whyman, S. Zacchini, Organometallics 21 (2002) 1832.
[13] W. Clegg, G.R. Eastham, M.R.J. Elsegood, B.T. Heaton, J.A. Iggo, R.P. Tooze, R.
Whyman, S. Zacchini, J. Chem. Soc. Dalton Trans. (2002) 3300.
[14] G. Cavinato, L. Toniolo, J. Organomet. Chem. 398 (1990) 187.
[15] A. Vavasori, G. Cavinato, L. Toniolo, J. Mol. Catal. 176 (2001) 11.
[16] G. Cavinato, A. Vavasori, L. Toniolo, F. Benetollo, Inorg. Chim. Acta 343 (2003)
183.
3.4.5. Proposed catalytic cycles
[17] G. Cavinato, L. Toniolo, A. Vavasori, J. Mol. Catal. A: Chem. 219 (2004) 233.
[18] G. Cavinato, A. Vavasori, L. Toniolo, A. Dolmella, Inorg. Acta 357 (2004) 2737.
[19] E. Amadio, G. Cavinato, A. Dolmella, L. Ronchin, L. Toniolo, A. Vavasori, J. Mol.
Catal. A: Chem. 298 (2009) 103.
[20] G. Cavinato, S. Facchetti, L. Toniolo, J. Mol. Catal. A: Chem. 333 (2010) 180.
[21] R.P. Tooze, K. Whiston, A.P. Malyan, J. Taylor, N.W. Wilson, J. Chem. Soc. Dalton
Trans. (2000) 3441.
[22] C. Bianchini, A. Meli, W. Oberhauser, P.W.N.M. van Leeuwen, M.A. Zuideveld, Z.
Freixa, P.J. Kamer, A.L. Spek, O.V. Gusev, M. Kal’sin, Organometallics 22 (2003)
2409.
[23] E. Drent, J.A.M. van Broekhoven, M.J. Doyle, J. Organomet. Chem. 417 (1991)
235.
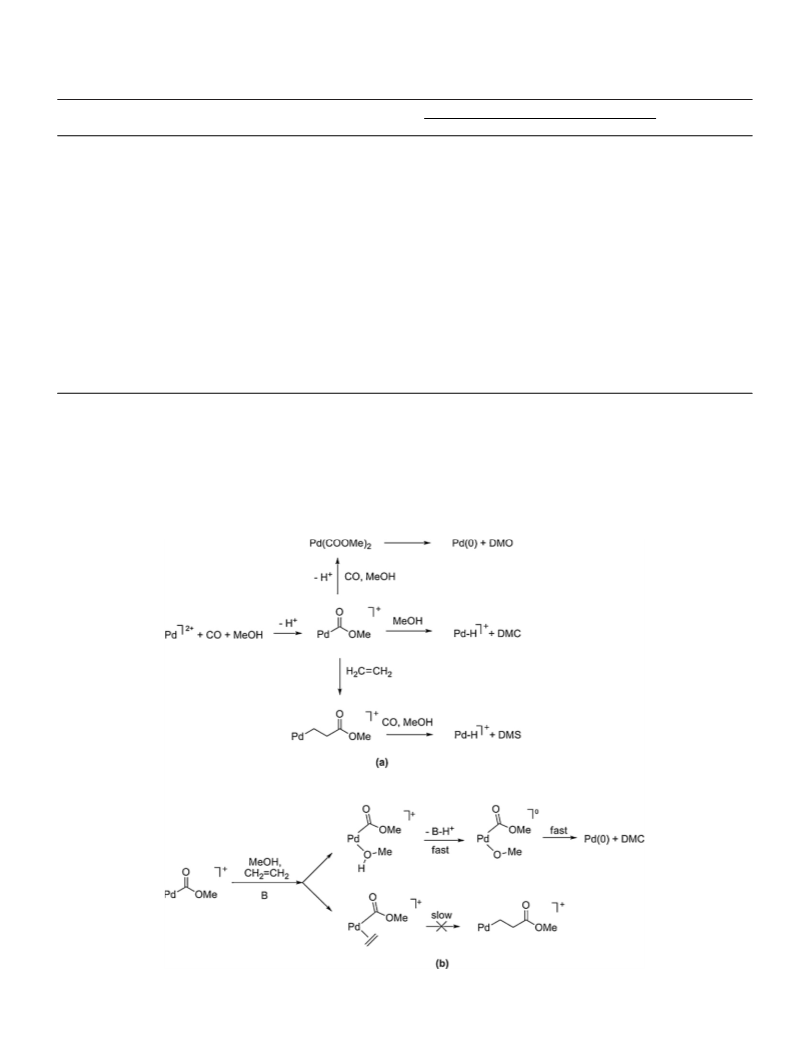
As already stated, in the absence of BQ, MP may form via the
“Pd–H” mechanism (reaction (3)). In the presence of BQ it may
form via either cycle 3 or 4. Fig. 4 shows the proposed catalytic
cycles for the formation of DMC, DMO and DMS. The one rele-
vant to DMO (cycle 2) proceeds via a typical Pd(II)/Pd(0) catalytic
cycle where a cis-dimethoxy intermediate yields the product and
a Pd(0)–BQ complex, whose oxidation is promoted by HX to a
Pd(II)–(OC6H4OH) intermediate, which reacts further to give H2BQ
this case HX is MeOH.
[24] E. Amadio, G. Cavinato, A. Dolmella, L. Toniolo, Inorg. Chem. 49 (2010) 3721.
[25] A.I. Min’kov, O.A. Efimov, N.K. Eremenko, Akademii Nauk SSSR, Seriya Khimich-
eskaya 5 (1989) 998.
In cycles 1 and 3 the methanolysis product-forming steps to
DMS or DMC are accompanied by the formation of a Pd–H species
[22], which is promptly consumed by BQ, probably with forma-
tion of a of a Pd(II)–(OC6H4OH) species [38]. These two catalytic
cycles do not occur through a Pd(II)/Pd(0) cycle species, as pro-
posed for cycle 2. In any case, all three are equivalent, because also
cycles 1 and 3 might occur through a Pd(0)–BQ intermediate, whose
oxidation would be promoted by HX as proposed mechanism 2.
[26] J.M. Jenkins, J.C. Verkade, Inorg. Chem. 11 (1968) 108.
[27] T.A. Stephenson, S.M. Morehouse, A.R. Powell, J.P. Heffer, G. Wilkinson, J. Chem.
Soc. (1965) 3362.
[28] G. Cavinato, L. Toniolo, J. Organomet. Chem. 444 (1993) C65.
[29] R. Bertani, G. Cavinato, L. Toniolo, G. Vasapollo, J. Mol. Catal. 84 (1993) 165.
[30] F. Rivetti, U. Romano, J. Organomet. Chem. 154 (1978) 323.
[31] F. Ramirez, S. Dershowitz, J. Am. Chem. Soc. 78 (1956) 5614.
[32] M. Arshad, A. Beg, M.S. Siddiquiri, Tetrahedron 22 (1966) 2203.
[33] W.P. Mul, H. Oesterbeek, G.A. Beitel, G.J. Kramer, E. Drent, Angew. Chem. Int.
Ed. 39 (2000) 1848.
[34] V.R. Khabibulin, A.V. Kulik, I.V. Oshanina, L.G. Bruk, O.N. Temkin, V.M. Nosova,
A. Yu Ustynyuk, V.K. Bel’skii, A.I. Stash, A.L. Lysenko, M. Yu Antipin, Kinet. Katal.
48 (2007) 228.
[35] M.A. Zuideveld, P.C.J. Kamer, P.W.N.M. van Leeuwen, P.A.A. Klusener, H.A. Stil,
C.F. Roobeek, J. Am. Chem. Soc. 120 (1998) 7977.
4. Conclusions
In summary, we have reported that Pd(II)–PPh3 and PdX2 (X = Cl,
Br, I) complexes, in combination with BQ, catalyze the oxidative
carbonylation of ethene in MeOH, yielding MP and DMS, together
with DMC and DMO. The formation of DMS unambiguously proves
that ethene inserts into a Pd–COOMe bond. BQ sweeps away Pd–H
[36] C. Bianchini, A. Meli, W. Oberhauser, J. Chem. Soc. Dalton Trans. (2003)
2627.
[37] P.W.N.M. van Leeuwen, M.A. Zuideveld, B.H.G. Swennenhuis, Z. Freixa, P.C.J.
Kamer, K. Goubitz, J. Fraanje, M. Lutz, A.L. Spek, J. Am. Chem. Soc. 125 (2003)
5523.
[38] H. Grennberg, A. Gogoll, J.E. Baeckvall, Organometallics 12 (1993) 1790.

 Cavinato, Gianni
Cavinato, Gianni