P.Y. Zhang et al. / Chinese Chemical Letters 21 (2010) 1307–1309
1309
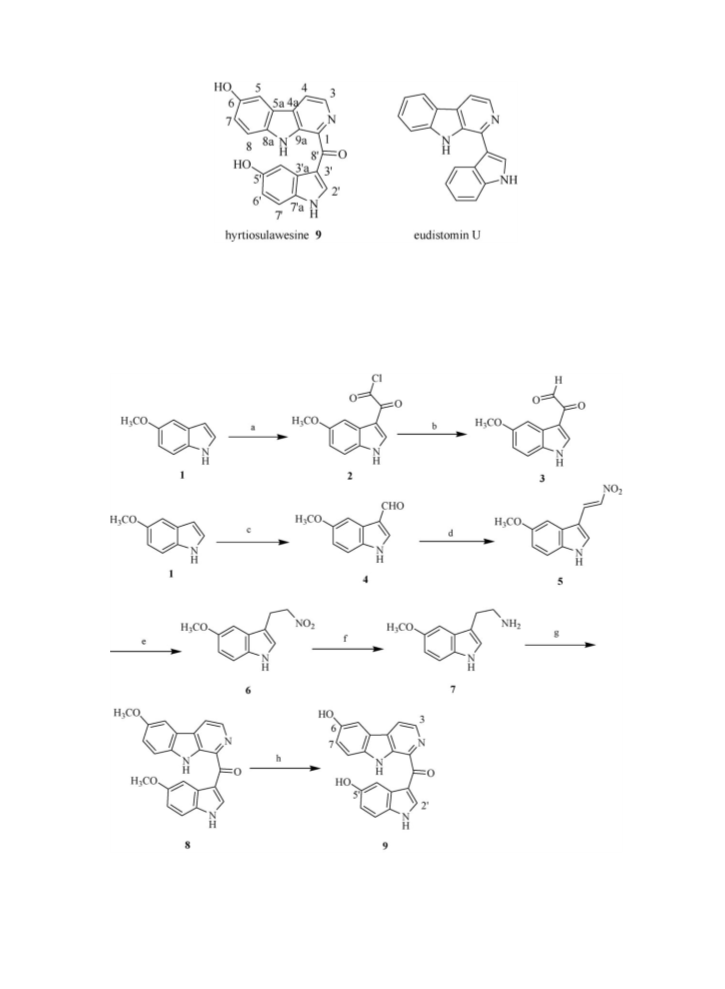
room temperature gave compound 6 in 98% yield. Subsequently, compound 6 was reduced by H2 in the presence of Pd/
C in methanol at room temperature to give 5-methoxy tryptamine 7 in 99% yield.
The classical method of preparing b-carboline alkaloids through Pictet–Spengler reaction was a two-step method,
and involved the acid-catalyzed condensation of an aliphatic amine attached to a sufficiently reactive aromatic nucleus
with aldehydes. Specifically, in the first step, an imine was formed, which can be activated by acids, while in the second
step, the endo cyclization occurred between a carbon nucleophile of a sufficiently reactive aromatic moiety and the
activated iminium ion, resulting in tetrahydro-b-carboline by the formation of N-heterocyclic ring through a new C–C
bond. After dehydrogenation, tetrahydro-b-carboline was converted to b-carboline [14–16].
Yang et al. have described a one-step conversion of L-tryptophon and activated aldehydes directly to 1-substituded
b-carbolines in the presence of acid under modified Pictet–Spengler reaction [17]. They explored the transformation
using H2SO4, p-TsOH, and HCl as acids, different solvent systems (MeOH, acetone, MeCN, THF, 1,4-dioxane,
DMSO, and DMF), reaction temperatures and reaction time. However, the methods of Yang’s report resulted in poor
yields of the desired target compounds [17]. In our experiments, the treatment of 5-methoxy tryptamine 7 with the
compound 3 in acetic acid directly generated a dehydrogenated b-carboline product 8 in one-pot in 40% yield. The
compound 8 was then transformed into the target compound hyrtiosulawesine 9 in the presence of BBr3 in CH2Cl2 at
ꢀ78 8C with the yields of 95%. The spectroscopic properties of the target compound 9 are found to be in good
agreement with those reported for the natural product [11]. The total synthetic route was showed in Scheme 1. All
1
structures of new compounds were confirmed by H NMR, 13C NMR and HRMS [18].
On the whole, the modified one-pot oxidation reaction was more efficient and convenient in preparing 1-substituted
b-carbolines without the need of aromatization step. It is highly expected that this methodology could be widely used
in the all b-carboline compounds. Further application of this strategy is underway in our laboratory.
Acknowledgment
We are grateful for the financial support of Key International S&T Cooperation Projects of MOST (No.
2008DFA31040).
References
[1] E. Magnier, Y. Langlois, Tetrahedron 54 (1998) 6201.
[2] A. Batch, R.H. Dodd, J. Org. Chem. 63 (1998) 872.
[3] R.S. Kusurkar, S.K. Goswami, S.M. Vyas, Tetrahedron Lett. 44 (2003) 4761.
[4] J. McNulty, I.W. Still, J. Curr. Org. Chem. 4 (2000) 121.
[5] Y.C. Song, J. Wang, S.F. Teng, et al. Bioorg. Med. Chem. Lett. 12 (2002) 1129.
[6] B.A. Shinkre, S.E. Velu, Synth. Commun. 37 (2007) 2399.
[7] A. Badre, A. Boulanger, E. Abou-Mansour, et al. J. Nat. Prod. 57 (1994) 528.
[8] G. Massiot, S. Nazabadioko, C. Bliard, J. Nat. Prod 58 (1995) 1636.
[9] P. Molina, P.M. Fresneda, S.G. Zafra, Tetrahedron Lett. 36 (1995) 3581.
[10] P. Rocca, F. Marsais, A. Godard, et al. Tetrahedron Lett. 36 (1995) 7085.
´ ´
[11] S. Mostafa, D. Christine, D. Desire, et al. J. Nat. Prod 65 (2002) 1173.
[12] C.A.B. Wager, S.A. Miller, J. Label. Compd. Radiopharm. 49 (2006) 615.
[13] M. Marco, R. Silvia, S. Claudia, et al. J. Med. Chem. 41 (1998) 3831.
[14] A. Pictet, T. Spengler, Ber. Dtsch. Chem. Ges. 44 (1911) 2030.
[15] E.D. Cox, J.M. Cook, Chem. Rev. 95 (1995) 1797.
[16] S. Duggineni, D. Sawant, B. Saha, et al. Tetrahedron 62 (2006) 3228.
[17] M.L. Yang, P.C. Kuo, A.G. Damu, et al. Tetrahedron 62 (2006) 10900.
[18] Spectral data. Compound 8 mp 220–221 8C; HRMS: m/z calcd. for C22H18N3O3+, 372.1348; found: 372.1341; 1H NMR (600 MHz, DMSO-
d6): d 12.00 (s, 1H), 11.88 (s, 1H), 8.52 (d, 1H, J = 4.6 Hz), 8.38 (d, 1H, J = 4.6 Hz), 8.10 (d, 1H, J = 2.3 Hz), 7.86 (d, 1H, J = 2.3 Hz), 7.75 (d,
1H, J = 8.7 Hz), 7.46 (d, 1H, J = 8.7 Hz), 7.23 (dd, 1H, J = 2.3, 8.7 Hz), 6.90 (dd, 1H, J = 2.3, 8.7 Hz), 3.88 (s, 3H), 3.85 (s, 3H); 13C NMR
(150 MHz, DMSO-d6): d 188.5, 154.7, 153.0, 139.5, 139.2, 138.0, 137.1, 136.1, 133.4, 132.6, 129.8, 122.6, 120.7, 118.7, 116.2, 114.1, 113.5,
108.0, 106.9. Compound 9 mp 362–363 8C; HRMS: m/z calcd. for C20H12N3O3ꢀ, 342.0879; found: 342.0875; 1H NMR (600 MHz, methanol-
d4): d 8.74 (s, 1H, H-20), 8.40 (d, 1H, J = 5.0 Hz, H-3), 8.20 (d, 1H, J = 5.0 Hz, H-4), 7.98 (d, 1H, J = 2.3 Hz, H-40), 7.57 (d, 1H, J = 2.3 Hz, H-
5), 7.53 (d, 1H, J = 8.7 Hz, H-8), 7.32 (d, 1H, J = 8.7 Hz, H-70), 7.16 (dd, 1H, J = 2.3, 8.7 Hz, H-7), 6.83 (dd, 1H, J = 2.3, 8.7 Hz, H-60); 13C
NMR (150 MHz, methanol-d4): d 188.5 (C-80), 154.7 (C-50), 153.0 (C-6), 139.5 (C-1), 139.2 (C-20), 138.0 (C-8a), 137.1 (C-9a), 136.1 (C-3),
133.4 (C-4a), 132.6 (C-70a), 129.8 (C-30a), 122.6 (C-4a), 120.7 (C-7), 118.7 (C-4), 116.2 (C-30), 114.1 (C-8, C-60), 113.5 (C-70), 108.0 (C-40),
106.9.(C-5).

 Zhang, Pu Yong
Zhang, Pu Yong