Y. Hu et al. / Tetrahedron: Asymmetry xxx (2015) xxx–xxx
5
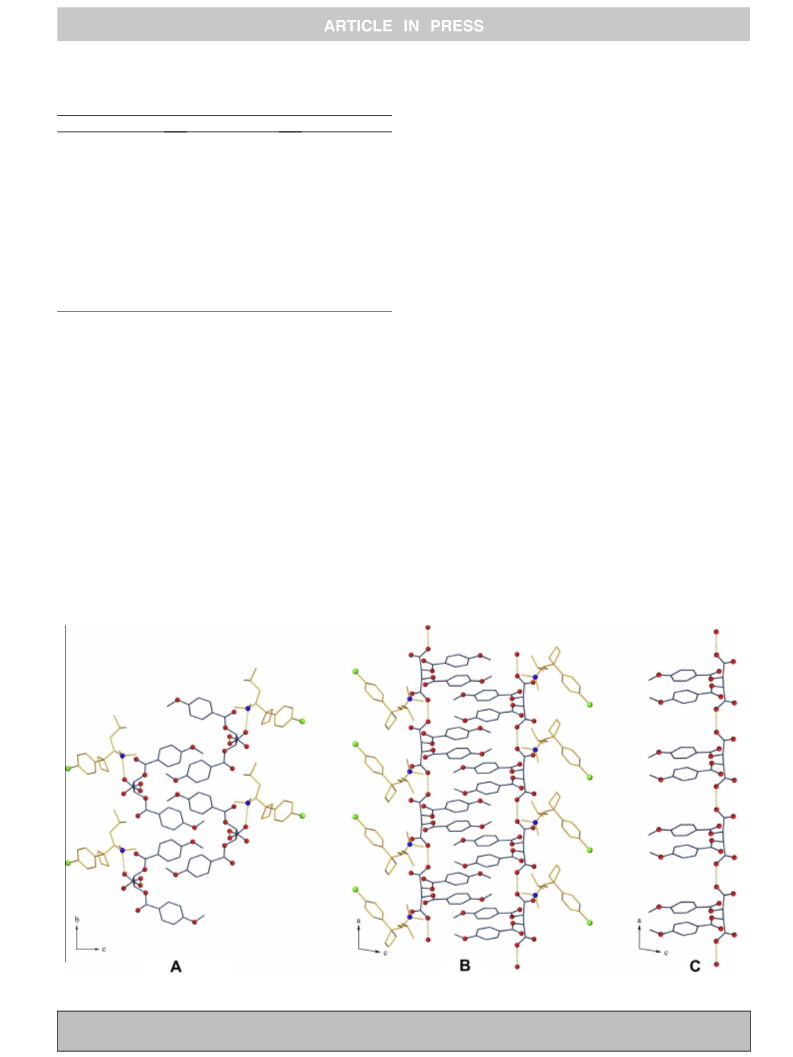
Figure 3. Crystal structures of less-soluble salt from crystal (R)-1ꢀ(S,S)-DBTA 7.9 (A) Top view and (B) side view down the screw axis of DBTA; (C) helix structure of (S,S)-DBTA
molecules.
functional groups and configuration of the drugs. The resolution
process is a simple model for recognition, which plays an important
role in biological systems and the preferred ‘biological answer’ is
the crystallization of one of the diastereomeric salts.
with ethyl acetate (300 mL ꢁ 4). The organic layer was combined
and washed with water (300 mL ꢁ 2). The organic phase was dried
over anhydrous Na2SO4. After the solvent was removed, the free
base of sibutramine (41.6 g, 99.4% yield) was obtained as a solid.
Mp: 53 °C; 1H NMR (400 MHz, CDCl3, d): 7.22 (d, J = 8.4 Hz, 2H),
7.14 (d, J = 8.4 Hz, 2H), 2.89 (dd, J = 2.3, 10.8 Hz, 1H), 2.47–2.40
(m, 1H), 2.33–2.26 (m, 1H), 2.16 (s, 6H), 2.12–2.05 (m, 1H), 1.97–
1.87 (m, 1H), 1.80–1.71 (m, 1H), 1.56–1.48 (m, 1H), 1.26–1.02
(m, 3H), 0.96 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 6.5 Hz, 3H) ppm.
Sibutramine 1 (31.0 g, 0.11 mol) was added to a solution of 4
(41.7 g, 0.1 mol) in ethanol (530 mL). While stirring, n-hexane
(500 mL) was added to the solution. The mixture was heated at
reflux for 1 h and then cooled to room temperature. The resulting
colorless crystals were collected by filtration and recrystallized in
isopropanol. 32.6 g (84.5% yield and 98.6% ee) of the salt of (R)-
4. Experimental
4.1. General
Racemic sibutramine hydrochloride was obtained from com-
mercial resource and was used without further purification. The
resolving agents (R,R)-DBTA 3, (R,R)-DTTA 4, (R,R)-DMTA 5, and
(S,S)-DMTA were commercially available or made in our lab-
oratory.20 All solvents were purified by standard procedures. All
chemicals were analytically pure. 1H NMR and 13C NMR spectra
were taken on a 400 MHz spectrometer. Chemical shifts of 1H
NMR are expressed in ppm with the residual signal of CDCl3 as an
internal standard (d = 7.26 ppm). Melting points were obtained on
a digital melting point apparatus and are uncorrected. IR spectra
were measured on a Nicolet MX-1 spectrometer by the KBr method.
Optical rotations of chiral compounds were measured on
Perkin–Elmer 341 automatic polarimeter. The enantiomeric excess
values of 1 were determined by HPLC analyses using an Agilent-
1100 instrument equipped with Chiralpak OD-H column
(4.6 ꢁ 250 mm). The mobile phase was hexane/isopropanol
(90:10) with 0.05% trifluoroacetic acid and flow rate was 1.0 mL/
min under detection wavelength of 222 nm. Retention time: (R)-1
7.0 min, (S)-1 12.8 min.
1ꢀ4 was obtained. Mp: 161 °C; IR (KBr):
2847, 2040, 1919, 1716, 1614, 1506, 1347, 1252, 1176, 1099,
1024, 846, 769, 699 and 521 cmꢂ1 1H NMR (400 MHz, CDCl3, d):
m = 3431, 2955, 2879,
;
8.03 (d, J = 8.8 Hz, 4H), 7.35 (d, J = 8.4 Hz, 2H), 7.26 (d, J = 8.4 Hz,
2H), 6.83 (d, J = 8.8 Hz, 4H), 5.92 (s, 2H), 3.82 (s, 6H), 3.48–3.45
(m, 1H), 2.58–2.19 (m, 10H), 1.74–1.69 (m, 2H), 1.63–1.58 (m,
1H), 1.44–1.36 (m, 1H), 1.28–1.22 (m, 1H), 1.01–0.96 (m, 6H)
ppm; 13C NMR (100 MHz, CDCl3, d): 169.4, 164.3, 162.4, 138.7,
132.4, 131.1, 128.9, 127.7, 121.3, 112.5, 70.8, 70.5, 54.4, 48.3,
34.3, 33.6, 32.5, 24.5, 22.0, 20.9, 14.8 ppm.
4.3. Preparation of salt (S)-1ꢀ4
(S)-1ꢀ(S,S)-DMTA (81.8% yield) was obtained from 1 and (S,S)-
DMTA by using the above mentioned method. The colorless solid
(6.97 g, 0.01 mol) was stirred in a solution of Na2CO3 (1.6 g,
0.015 mol) and then extracted with ethyl acetate (100 mL ꢁ 3).
The extracts were washed with water (100 mL ꢁ 2), dried over
4.2. Resolution of 1
The hydrochloride of sibutramine (47.3 g, 0.15 mol) was dis-
solved in a solution of Na2CO3 (20 g, 0.19 mol) and then extracted

 Hu, Yu
Hu, Yu