H. Cheropkina et al.
Biochemical Pharmacology 193 (2021) 114763
After the initial stopped-flow experiments of Beaty and Ballou more than
affinity and turnover numbers in presence/absence of well-known sub-
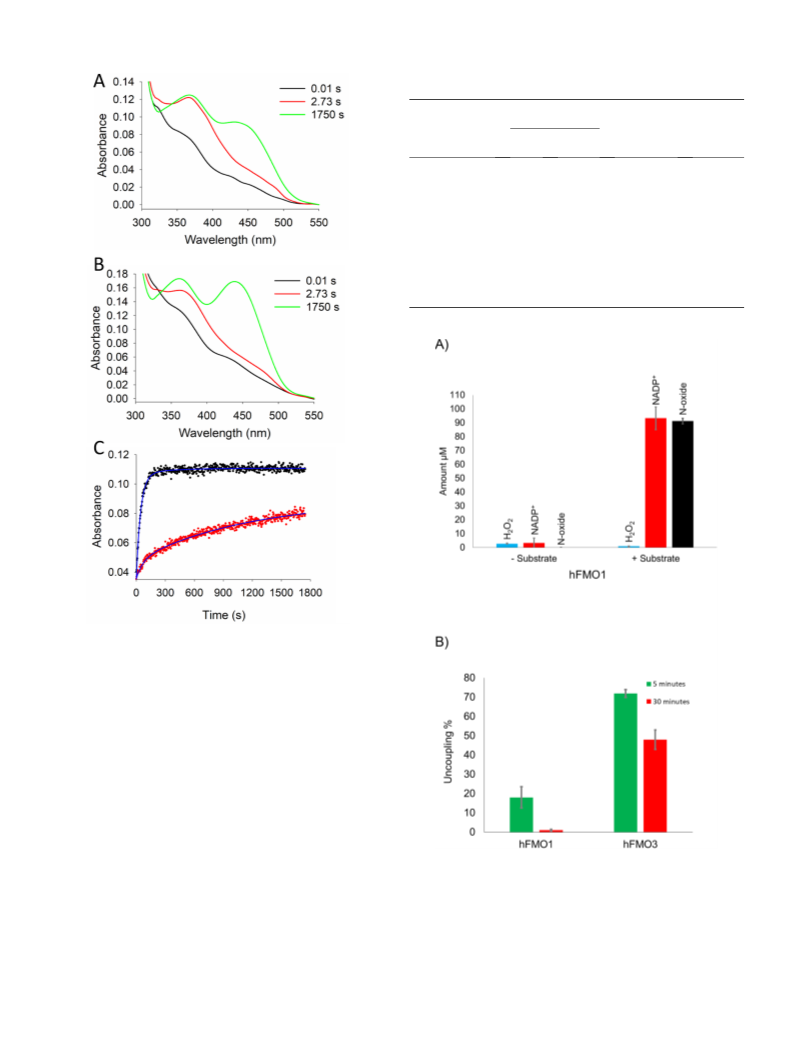
strates. The formation and decay of the C4a-hydroperoxyflavin inter-
mediate was monitored in the absence or presence of the physiological
substrate taurine. Finally, the uncoupling reactions of hFMO1 were
monitored using tamoxifen as a substrate comparing the amount of
oxidized NADPH to the amount of N-oxide product formed in the reac-
tion. The results should provide a comprehensive model for hFMO1 in
terms of structure–function relationship indicating a strategic role of
4
0 years ago, which led to the identification of the important interme-
diate in the catalytic cycle of these flavo-enzymes [11], the C4a-
hydroperoxyflavin for pig FMO1, this stable intermediate has been
only inferred for human FMO1 and never observed spectroscopically.
Only recently, AncFMO1 was tested in pre-steady state conditions, but
the intermediate was unstable and its formation could not be followed
[
16].
Dr. Daniel Ziegler’s lab was the first to determine the catalytic
mechanism of the first mammalian FMO, pig liver FMO, during 1970s to
990s [17]. Later, Beaty and Ballou provided the first evidence of the
+
NADP in providing stabilization to the reaction intermediate and a low
level of uncoupling. To this end, this work reports on the investigation of
the formation and the stability of the C4a-hydroperoxyflavin interme-
diate of hFMO1 by stopped-flow spectroscopy and the consequences for
the catalytic activity of this enzyme.
1
full reaction mechanism of FMOs in the 1980s [10,11]. In a remarkable
reference work for the field, it was shown how pFMO1 was initially
reduced by NADPH followed by binding of molecular oxygen and sta-
bilization of a long-lived hydroperoxy-intermediate [11] that still today
is ascribed as the moiety responsible for catalysis.
2. Materials and methods
As mentioned above, the stopped-flow experiments of Beaty and
Ballou were performed using the pFMO1 [11], but five different FMO
genes are present in humans [18,19]. The most relevant isoforms for
drug metabolism are FMO1 and FMO3 that are also polymorphic
2.1. Chemicals
Chromatographic resins were purchased from GE Healthcare (Italy).
Chemicals including Luria Bertani (LB), tryptone, agar, ampicillin, Iso-
propyl-beta-D-thiogalactopyranoside (IPTG), phenylmethanesulfonyl
fluoride (PMSF), riboflavin, flavin adenine dinucleotide (FAD), β-mer-
captoethanol, glycerol, lysozyme, NADPH, NADP , NADH, NAD ,
hypotaurine, taurine, tamoxifen, fenthion, fenthion sulfoxide, phos-
phoric acid, acetonitrile, methanol, triethylamine and salts were all
purchased from Sigma-Aldrich. Tamoxifen N-oxide standard was pur-
chased from Toronto Research Chemicals (Canada). All chemicals were
of highest available purity and used without any further purification. All
media, solutions and buffers were prepared with deionized Milli-Q
water.
[
20–23]. In addition, FMO5 is also expressed in the liver and some
recent publications suggest that it is an important drug metabolizing
enzyme capable of carrying out atypical Baeyer-Villiger oxidations in
addition to soft nucleophilic heteroatom monooxygenation [24,25]. In
the last 40 years bioinformatics and recombinant DNA technology has
allowed for the designing of clones and the subsequent heterologous
expression of FMOs in bacterial hosts such as E. coli [26]. More recently
it has been shown that hFMO3, that for a long time was thought to
behave like pFMO1, only transiently binds the substrate [27]. Stopped-
flow experiments demonstrated that hFMO3 forms and stabilizes only a
small amount of intermediate in the presence of oxygen that rapidly
+
+
+
decays [28]. A role for NADP in the stabilization of the intermediate
2.2. Protein expression and purification
and the overall structure of this enzyme was also demonstrated [28,29].
The real physiological substrate of hFMO1 was only discovered
The hFMO1 gene and the oligonucleotides encoding its amino- and
carboxy-termini were synthesized by GeneScript Biotech (Netherlands).
The gene was sub-cloned in pJL2 expression vector within the XbaI-
1
recently. The research group of Shephard used metabolite analysis by H
NMR spectroscopy to decipher the role of hFMO1 in the biosynthetic
pathway of taurine using hypotaurine as its substrate [30]. Taurine, a
highly abundant amino acid in humans, can be synthesized de novo from
hypotaurine. Furthermore, they also demonstrated the pH dependent
ability of hFMO1 to use either NADH or NADPH as reducing cofactor
′
HindIII restriction sites [33,34]. A stretch of four histidine residues (5
′
CACCATCACCAT 3 ) was inserted at the C-terminus to assist in the
purification.
The full-length hFMO1 protein was expressed in Escherichia coli
JM109 cells in two-liter conical flasks containing 500 mL of Terrific
[
30].
As the chemical nature of the C4a-hydroperoxyflavin intermediate is
◦
Broth (TB). Cells were induced by 1 mM IPTG and grown at 24 C with
ampicillin (100 g/mL), 50 mg/L of riboflavin. After 24 h post-induction
the same in all the FMO enzymes, its lifetime and therefore reactivity is
modulated by the protein environment. Two different approaches can be
followed; (1) resolving the crystal structure and looking at the protein
matrix surrounding the flavin cofactor to justify any differences
observed in the catalytic activity of different FMOs and/or (2) investi-
gate the formation and decay of the FAD intermediate by spectroscopy.
Regarding the first option, to date no 3D structure of any human FMO
has been resolved since they are membrane-bound proteins and not
stable in aqueous environment [31]. Association to membrane is a well-
known hindrance to the general crystallization process of proteins. To
overcome this obstacle, synthetic genes coding for ancestral FMO iso-
forms were recently synthesized and the resulting synthetic proteins
were crystalized leading to the structures of AncFMO2, AncFMO3-6,
AncFMO5 and AncFMO1 [16,32]. In this work the second option was
followed in order to investigate the reactivity of this intermediate in
hFMO1 and compare its behaviour to other human FMOs as well as its
homologous counterpart, pFMO1 (83.1% amino acid similarity). To
achieve this goal hFMO1 was expressed heterologously in E. coli and
purified by affinity chromatography. The secondary structure of the
purified protein was investigated by circular dichroism and the stabi-
μ
◦
cells were harvested by 20 min centrifugation at 4000 rpm at 4 C. The
◦
cell pellet was stored at ꢀ 20 C until use.
The protein was purified from the membrane fractions via DEAE
anion-exchange and Ni-chelating sepharose fast-flow affinity column,
following the procedure described previously for FMO3 [35]. Briefly,
pellet containing hFMO1 was resuspended in Buffer A (50 mM KPi pH
8.0, 20 % glycerol, 5 mM β-mercaptoethanol) with 0.5 mM PMSF using
5 mL/g of cells. This was followed by the addition of 0.5 mg/ml of
◦
lysozyme and the solution was stirred for 1 h at 4 C. The cells were then
lysed by ultrasonication (10 × 30 s). The resulting lysate was ultra-
◦
centrifuged at 41,000 rpm for 60 min at 4 C. Subsequently, the pellet
containing the membrane fraction was resuspended using Dounce ho-
◦
mogenizer in Buffer A with 1 % IGEPAL and left stirring for 2 h at 4 C.
This was followed by a second ultracentrifugation step (41,000 rpm for
◦
60 min at 4 C) after which the supernatant was loaded on a DEAE-
Sepharose Fast Flow column connected to the affinity chromatography
column. Human FMO1 protein was eluted with 40 mM histidine in 50
mM KPi pH 8.0, 0.1% IGEPAL and 20% glycerol. The elution profile was
monitored by UV/Vis spectroscopy and fractions containing the flavo-
protein were pooled and buffer exchanged (100 mM KPi pH 8.0, 20%
glycerol) using 30 kDa cutoff Amicon™ Ultra Centrifugal Filter. The
+
+
lizing role of NAD and NADP cofactors were determined by differ-
ential scanning calorimetry (DSC). These reduced cofactors were also
used in pre-steady state studies to assess their ability to efficiently
reduce the enzyme and further in steady-state studies to establish
◦
purified protein was stored at ꢀ 80 C in small aliquots to prevent
repeated thawing/freezing.
2

 Catucci, Gianluca
Catucci, Gianluca