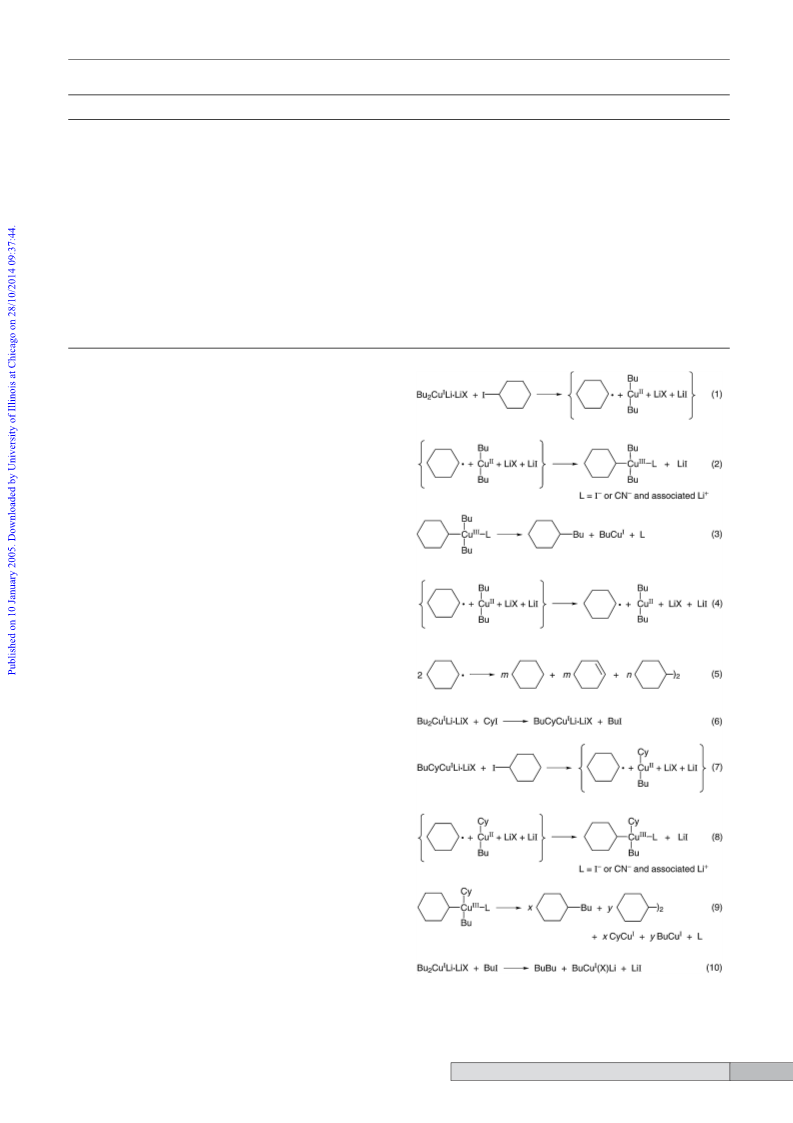
ˆ
Another possible source of BuBu is reductive elimination from
the ‘free’ Bu2CuII that results when Cy• escapes from the initial
solvent cage (step 4);1 however, theoretical calculations suggest
that this pathway is not energetically favorable,12 which also
means that BuCy does not come from BuCyCuII (step 7).
In light of Scheme 1, we can calculate the yields of products
that arise via transmetallation with the aid of eqns. 11–14, where
y(P) is the yield of product P, YR(P) is the yield of P from radical
routes that do not involve transmetallation, and YT(P) is the
yield of P from routes that involve transmetallation. In the latter
case transmetallation may be followed by radical reactions.
3%, vide supra). However, based on the corrected values of CCy
after 4 s (27 6% vs. 2.5 1.7%, vide supra), 1a appears to be
substantially more reactive.
True reactivity depends upon kinetic data, which our results
only approximate; nevertheless, it can be stated unequivocally
that 1b is not remarkably more reactive than 1a, as has been
asserted.17 The reactivity patterns of 1a and 1b are now in
harmony with the theoretical,18 spectroscopic,19 and X-ray
investigations,20 which concluded that both cuprates are in fact
varieties of the Gilman reagent.21
These results make it clear that scrupulous attention to exper-
imental detail is necessary for both mechanistic and synthetic
organocopper chemistry and that statistical tests are essential.
As a general precaution, all surfaces that come in contact with
organocuprates should be either virgin or passivated glass or an
inert material such as polypropylene, and the ages of solvent
stills and lots of Cu(I) salts should be scrutinized.
YR(Cy(−H)) = y(Cy(−H))
YR(CyH) = y(Cy(−H))
YR(CyCy) = 1.5 y(Cy(−H))
YT(P) = y(P) − YR(P)
(11)
(12)
(13)
(14)
Acknowledgements
Eqn. 11 originates with the observation that Cy(−H) only
appears in step 5 and the assumption that there is no other
source of this product. Based on the classic study of the
thermal decomposition of BuCuI by Whitesides et al.,15 thermal
decomposition of CyCuI would be expected to produce Cy(−H)
via b-hydride elimination and CyH via reaction of the HCuI
co-product with CyCuI. b-Hydride elimination in cuprates is
negligible at −78 ◦C,16 so that this assumption is a good one. It
is important to note that BuBu does not result from the thermal
decomposition of BuCu or Bu2CuLi.15,16
The authors thank J. P. Snyder for helpful discussions concerning
his calculations and J. K. Kochi for the passivation procedure.
The University of North Carolina–Charlotte, the Oak Ridge
Associated Universities (ORAU) and the US National Science
Foundation (NSF) provided financial support.
References
1 S. H. Bertz, G. Dabbagh and A. M. Mujsce, J. Am. Chem. Soc., 1991,
113, 631–636.
2 (a) W. A. Cramer, J. Phys. Chem., 1967, 71, 1171–1174 (kd/kc
Eqn. 12 follows from the stoichiometry of step 5, in which
equimolar amounts of CyH and Cy(−H) are formed. Eqn. 13 is
derived from eqns. 11 and 12 and the experimentally determined
ratio of disproportionation to combination for cyclohexyl
radicals, kd/kc = [YR(CyH) + YR(Cy(−H))]/YR(CyCy) = 1.3.2
Eqn. 14 results from the fact that the two routes represented
by YR(P) and YT(P) are mutually exclusive as defined, and thus
y(P) = YR(P) + YT(P).
=
1.1); (b) J. W. Falconer and M. Burton, J. Phys. Chem., 1963, 67,
1743–1746 (kd/kc = 1.5); (c) We use the mean, kd/kc = 1.3.
3 G. M. Whitesides, W. F. Fischer, Jr., J. San Filippo, Jr., R. W. Bashe
and H. O. House, J. Am. Chem. Soc., 1969, 91, 4871–4882.
4 Confidence limits are calculated at the 95% confidence level, unless
otherwise noted; see L. L. Havlicek and R. D. Crain, Practical
Statistics for the Physical Sciences, American Chemical Society,
Washington D.C., 1988.
5 H. O. House, C.-Y. Chu, J. M. Wilkins and M. J. Umen, J. Org.
For the eight reactions of 1a in Table 1, the average yields (%)
are y(BuCy) = 58.8 1.3, y(CyH) = 9.4 1.3, y(Cy(−H)) =
3.8 0.5, y(CyCy) = 9.1 1.7 and y(BuBu) = 5.9 2.5. Then,
we have YR(CyH) = 3.8, YR(CyCy) = 5.7, YT(CyH) = 5.6 and
YT(CyCy) = 3.4. Since YT(BuCy) = YT(CyCuI) = YT(CyH), we
can calculate the selectivity S of the reductive elimination in step
9 as S = YT(BuCy)/YT(CyCy) = 5.6/3.4 = 1.6. This result is
reasonable, given that the steric hindrance in the transition state
is higher for CyCy than BuCy.
Chem., 1975, 40, 1460–1469.
6 K. Tennakone, G. R. R. A. Kumara, I. R. M. Kottegoda, V. P. S.
Perera, G. M. L. P. Aponsu and K. G. U. Wijayantha, Sol. Energy
Mater. Sol. Cells, 1998, 55, 283–289.
7 P. Tomboulian, D. Amick, S. Beare, K. Dumke, D. Hart, R. Hites,
A. Metzger and R. Nowak, J. Org. Chem., 1973, 38, 322–325.
8 (a) T. M. Burkat and D. P. Dobychin, Fiz. Khim. Stekla, 1992, 18,
129–140; (b) M. Shimbo, Proc. 10th Int. Congr. Glass, 1974, 9, 7–14.
9 S. H. Bertz, A. Chopra, M. Eriksson, C. A. Ogle and P. Seagle, Chem.
Eur. J., 1999, 5, 2680–2691.
For the eight reactions of 1b in Table 1, the average yields (%)
are y(BuCy) = 81.3 3.9, y(CyH) = 6.5 1.5, y(Cy(−H)) =
10 B. H. Lipshutz and R. S. Wilhelm, J. Am. Chem. Soc., 1982, 104,
4696–4698.
1.2
0.2, y(CyCy) = 5.1
1.1 and y(BuBu) = 5.9
1.0.
11 J. P. Collman and L. S. Hegedus, Principles and Applications of
Organotransition Metal Chemistry, University Science Books, Mill
Valley, CA, 1980, pp. 544–565.
12 R. Rodebaugh, J. S. Debenham, B. Fraser-Reid and J. P. Snyder,
J. Org. Chem., 1999, 64, 1758–1761.
13 See also: E. J. Corey and G. H. Posner, J. Am. Chem. Soc., 1968, 90,
Then, we have YR(CyH) = 1.2, YR(CyCy) = 1.8, YT(BuCy) =
YT(CyH) = 5.3 and YT(CyCy) = 3.3. The selectivity of reductive
elimination in step 9 is S = YT(BuCy)/YT(CyCy) = 5.3/3.3 =
1.6. The agreement between the values of S for 1a and 1b gives
us confidence that our mechanistic scheme is essentially correct,
as L would not be expected to exert a significant steric influence.
The amount of CyH from transmetallation is approximately
the same for 1a and 1b. In contrast, the amount of Cy(−H)
from 1b is one-third of that from 1a. The main reason for the
lower yields of BuCy from 1a is the increased role of radical side
5615–5616.
14 (a) S. H. Bertz and C. P. Gibson, J. Am. Chem. Soc., 1986, 108, 8286–
8288; (b) See also: G. M. Whitesides, J. San Filippo, Jr., C. P. Casey
and E. J. Panek, J. Am. Chem. Soc., 1967, 89, 5302–5303.
15 G. M. Whitesides, E. R. Stedronsky, C. P. Casey and J. San Filippo,
Jr., J. Am. Chem. Soc., 1970, 92, 1426–1427.
16 S. H. Bertz and G. Dabbagh, J. Chem. Soc., Chem. Commun., 1982,
1030–1032.
17 B. H. Lipshutz, R. S. Wilhelm and D. M. Floyd, J. Am. Chem. Soc.,
ˆ
reactions. The corrected mean cyclohexyl conversions CCy (%)
are (81.1 3.8)/0.90 = 90 4 for 1a and (94.0 3.0)/0.99 =
95 3 for 1b, whereas the renormalized butyl conversions (%)
are 78 6 and 92 4, respectively (vide supra).
1981, 103, 7672–7674.
18 J. P. Snyder and S. H. Bertz, J. Org. Chem., 1995, 60, 4312–4313.
¨
19 (a) S. H. Bertz, K. Nilsson, O. Davidsson and J. P. Snyder, Angew.
After 1 h, the reactions of both 1a and 1b have reached
plateaus, and from the corrected yields of BuCy, (58.8
1.3)/0.90 = 65 1 vs. (81.3 3.9)/0.99 = 82 4, respectively, the
latter appears to be much more reactive. While one compound
may be the ‘desired product’, all the products must be considered
when discussing ‘reactivity’. Then, the latter appears to be only
Chem., Int. Ed., 1998, 37, 314–317.
20 (a) G. Boche, F. Bosold, M. Marsch and K. Harms, Angew. Chem.,
Int. Ed., 1998, 37, 1684–1686; (b) C. M. P. Kronenburg, J. T. B. H.
Jastrzebski, A. L. Spek and G. van Koten, J. Am. Chem. Soc., 1998,
120, 9688–9689.
21 For a succinct review, see N. Krause, Angew. Chem., Int. Ed., 1999,
38, 79–81.
ˆ
slightly more reactive after 1 h, based on CCy (90 4% vs. 95
3 9 4
O r g . B i o m o l . C h e m . , 2 0 0 5 , 3 , 3 9 2 – 3 9 4

 Bertz, Steven H.
Bertz, Steven H.