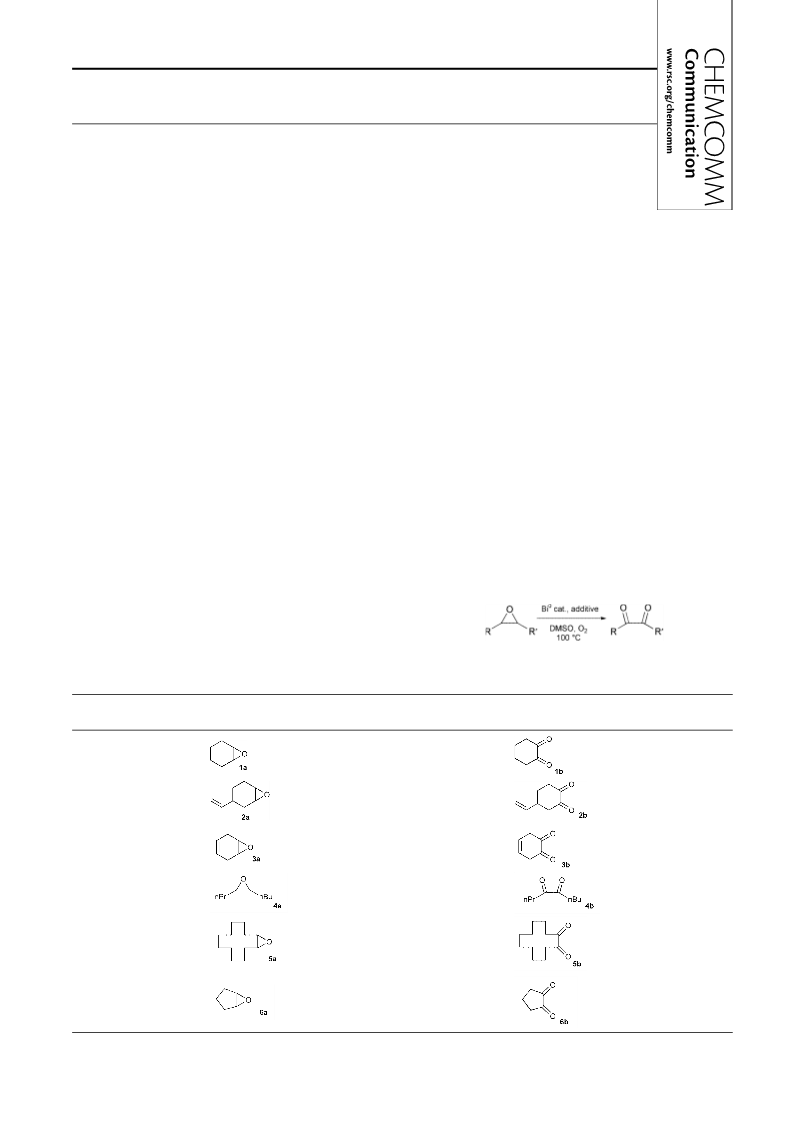
spectively), issued from the epoxide isomerisation in acidic
medium.8 No other by-products were detected.
The oxidation of unsaturated epoxides 2a and 3a indicated
that olefin functions were not modified by the catalytic
system.
oxygen at the pressure of 1 atm as the oxidant in this step is an
interesting aspect of the process. Recently, another Bi(III)–O2
system has proven its efficiency in the deprotection of S,S-
acetals.10
In conclusion, a new synthetic transformation is described,
involving the oxidative ring opening of epoxides to a-
diketones, in a combined Bi(0)–Cu(OTf)2 catalytic system
under O2–DMSO. The process uses commercially available
reagents in a one-pot reaction under relatively mild condi-
tions.
The a-diketones in five or six-membered rings were mainly
1
found in their enol form according to H-NMR analysis (in
CDCl3 at 20 °C). Particularly in the case of entry 3, only the
1,2-dihydroxybenzene form was observed. However, diketones
4b and 5b were present with low enolisation (respectively 36
and 30%) under the same analytical conditions.
S. A. thanks the French Ministry of Research for a doctoral
fellowship.
When a terminal epoxide such as 1-octene oxide was treated
under the Bi(0)–Cu(OTf)2 oxidation conditions (as for entry 1),
the expected a-keto aldehyde underwent further C–C cleavage
leading to a mixture of heptanal and heptanoic acid in 80%
overall yield. This result was consistent with our previous
observations on the reactivity of terminal epoxides.6
The presence of Bi(0) was essential for the preparation of the
a-diketone. In the absence of bismuth, 1a did not lead to 1b but
afforded 2-hydroxycyclohexanone in 55% yield. On the other
hand, no diketone was obtained in the absence of O2 (reactions
under N2) or in the absence of DMSO (reactions in DMF). This
result indicates that DMSO would act as the oxygen transfer
agent to the epoxide. Accordingly, dimethyl sulfide is evolved
in the oxidation process. It was also shown that without any
additive, the epoxide was unreactive.
Notes and references
General procedure for epoxide oxidation. A mixture of bismuth(0) (0.5
mmol) and the additive (0.5 mmol) in DMSO (15 ml) is heated at 100 °C
under O2 (1 atm). The epoxide (5mmol) in DMSO (5 ml) is then introduced
through a serum cap and the mixture is stirred at this temperature until
complete consumption of the epoxide (monitored by GC). The reaction
mixture is hydrolysed with brine (50 ml) and extracted with diethyl ether (3
3 50 ml). The combined organic layers are dried over MgSO4 and
evaporated. The product is purified by column chromatography over silica
1
gel with dichloromethane as the eluent. The products are identified by H
and 13C NMR and mass spectroscopy, and compared with authentic
commercial samples or literature data.
In order to check if Bi(III) species were involved in the
catalytic cycle, the reaction of 1a was carried out in the presence
of Bi(III)-mandelate,9 replacing Bi(0) (conditions of entry 1).
The reaction also afforded 1b, but in low yield (10%). This
result was taken to indicate that the reaction could proceed
through a Bi(0)/Bi(III) redox couple, in agreement with recent
results.6 We observed that the Bi(0) oxidative dissolution
(presumably to Bi(III)) needed molecular oxygen as well as the
presence of the epoxide and the additive. Under nitrogen, the
black Bi(0) precipitate persisted, and the epoxide oxidation to
the a-diketone did not take place.
1 (a) S. Rampalli, S. S. Chaudhari and K. G. Akamanchi, Synthesis, 2000,
1, 78; (b) V. Kesevan, D. Bonnet-Delpon and J. P. Bégué, Tetrahedron
Lett., 2000, 41, 2895; (c) S. E. Denmark, P. A. Barsanti, K.-T. Wong and
R. A. Stavenger, J. Org. Chem., 1998, 63, 2428; (d) S. Matsunaga, J.
Das, J. Roels, E. M. Vogl, N. Yamamoto, T. Iida, K. Yamaguchi and M.
Shibasaki, J. Am. Chem. Soc., 2000, 122, 2252.
2 T. M. Santusso and D. Swern, J. Org. Chem., 1975, 40, 2764.
3 S. C. Roy and S. Adhikari, Indian J. Chem., 1992, 31B, 459.
4
T. Zevaco, E . Duñach and M. Postel, Tetrahedron Lett., 1993, 34,
2601.
5 For catalytic oxidation of other substrates to a-diketones, see for
example: (a) F. A. Kham, B. Prabhudas, J. Dash and N. Sahu, J. Am.
Chem. Soc., 2000, 122, 9558; (b) C. M. Amon, M. G. Banwell and G. L.
Gravatt, J. Org. Chem., 1987, 52, 4851.
6 C. Coin, V. Le Boisselier, I. Favier, M. Postel and E. Duñach, Eur. J.
Org. Chem., 2001, 735.
7 N. Irwing-Sax and R. J. Bewis, in Dangerous properties of industrial
materials, Van Nostrand Reinhold, New York, 1989, 283.
8 (a) B. C. Ranu and U. Jana, J. Org. Chem., 1998, 63, 8212; (b) A. M.
Anderson, J. M. Blazek, P. Garg, B. J. Payne and R. S. Mohan,
Tetrahedron Lett., 2000, 41, 1527.
9 T. Zevaco and M. Postel, Synth. React. Inorg. Met.-Org. Chem., 1992,
22, (2&3), 289.
For the reaction mechanism, still under investigation, we
propose the initial oxidative oxirane ring opening to an a-
hydroxy ketone intermediate catalysed by the system Cu(OTf)2
[or TfOH]–DMSO. The presence of a strong acid (Lewis or
protic one) to activate the oxirane ring seems an important
feature of this process. The presence of weakly coordinating
triflate species could also have an important role in the reaction.
DMSO, in association to the acid, effects the oxidative ring
opening.
In a second step, in the presence of Bi(III), presumably issued
from Bi(0) oxidative dissolution, a redox reaction affords the a-
diketone by oxidation of the ketol. The reduced bismuth species
are reoxidized to Bi(III) under molecular oxygen. The use of
10 N. Komatsu, A. Taniguchi, M. Uda and H. Suzuki, Chem. Commun.,
1996, 1847.
Chem. Commun., 2001, 2566–2567
2567

 Antoniotti
Antoniotti