Inorganic Chemistry
Article
the product mixture again, followed by evaporation with liquid N2
previous paragraph and in entry 4 in Table 1, the solid mixture
containing the urea product was dried under vacuum for 30 min to 1
h to remove the water. The sample was then ground to a fine powder
and stirred using a spatula to obtain a uniform sample. The solid after
drying and grinding has a total mass of 1440 mg. A 42.6 mg portion of
trap at 50 °C for another 30 min. Water in 5−10 mL was added to
dissolve the solid, and CO was vigorously bubbled into the solution
2
for 3 min to precipitate copper(II) in the form of the basic copper
carbonate (Cu CO (OH) ). The cyan solid was filtered out and dried
2
3
2
on a suction funnel at 23 °C for 30 min.
the total sample was dissolved in 0.75 mL DMSO-d and analyzed by
6
1
13
Catalytic Urea Formation by the Recovered Basic Copper
H{ C} NMR spectroscopy.
Carbonate. The recovered basic copper carbonate (0.128 mmol or
Benzene, an internal standard in 8.9 mg, was then added to the
2
8 mg of Cu CO (OH) ) was used as a catalyst in 1% loading, based
1
13
2
3
2
DMSO-d solution. The solution was analyzed using H{ C} NMR
6
on copper(II), for urea synthesis in 0.5 mL of water at 120 °C over 15
spectroscopy with the six equivalent proton signals of benzene,
referenced at 7.33 ppm. The broad chemical shift at 5.49 ppm was
assigned to the four N−H equivalent hydrogen atoms of urea. The
urea was detected at a 24% yield (6.2 mmol) based on the starting
ammonium carbamate material (26 mmol). No significant amount
h. Urea formed in a 2% yield, which was measured using quantitative
1
13
H{ C} NMR spectroscopy with benzene (15 mg) as the internal
standard. This reaction was carried out in an open glass vial (1.18 ×
.87 in, made by MaxMau) that was placed in a C-276 Hastelloy
0
reactor to prevent corrosion from the metal reactor. Only the urea
formed inside the glass vial was analyzed for the yield. Under
otherwise identical conditions, Complex 1 gave urea in a 4% yield.
UV−Vis Quantitation of the Recovered Complex 1. To
quantify the recovered complex, the reaction, described in entry 1 in
Table 1 was performed again. At the end of the reaction, water, urea,
and ammonium carbamate were removed by drying in vacuum,
methanol extraction (2 × 15 mL), and centrifugation, as described in
the previous paragraph. The solid that is insoluble in methanol was
dissolved in a 1 M aqueous ammonia solution to form a 10 mL
solution using a volumetric flask. The absorption at λmax = 605 nm
was used to quantify Complex 1. A calibration plot was made using
(
>1%) of any other organic product was detected. The experiment
was conducted thrice to obtain an average yield of 18 ± 6%. The
characterization and quantitation methods were validated by testing a
purchased sample of urea in 40 mg.
Catalytic Urea Synthesis Using Complex 1 without Water at
50 °C (Entry 16 in Table 1). Complex 1 in the 0.2% catalyst
1
loading (12 mg, 0.051 mmol) and ammonium carbamate in 2.0 g (26
mmol) were added to a high-pressure metal reactor under air. An
oven was preheated to 150 °C. The oven door was briefly opened, a
high-pressure reactor was placed in the oven, and the oven door was
shut quickly. The temperature in the oven dropped to 125−130 °C
after placing the reactor, and it took 1 h for the temperature to
increase to 150 °C. The heating lasted for 17 h, the oven was turned
off, and the door was opened to allow cooling by air. The cooling took
1
0 mL solutions, which contained Complex 1 in 0.008, 0.016, 0.024
M, and a blank sample, which contained only a 1 M aqueous
−
1
−1
ammonia solution. The molar absorptivity was 53.6 L mol cm ,
and the path length was 1 cm. The concentration of the recovered
Complex 1 was 0.0073 M, corresponding to a 29% recovery yield.
The recovery yield of Complex 1 can be increased to 70% by the
following procedure. At the end of the reaction described in entry 1 in
Table 1, the product mixture was transferred to a round-bottom flask
with the help of 5 mL of methanol. Water in this solution was
3
0 min to 1 h for the reactor to reach an ambient temperature (25−50
°
C). The solid mixture containing the urea product was then dried in
vacuum for 30 min to 1 h to remove water, a side product.
The solid sample was then ground to a fine powder, which was
stirred using a spatula to obtain a uniform sample. After drying and
grinding, the solid has a total mass of 1147 mg, of which 40 mg of the
1
13
sample was analyzed via H{ C} NMR spectroscopy (see the
Quantitative Analysis of Urea Using Solution-Phase 1H{13C} NMR
Spectroscopy section for details). Urea was detected at a 15% yield
thoroughly removed under vacuum at 50 °C with a liquid N trap for
2
1
h. The resulting solid underwent methanol extraction (2 × 15 mL)
and centrifugation to remove urea from Complex 1, as described in
the Catalyst Recovery and Spectroscopic Analyses section. No
insoluble white solid was observed in this extraction procedure.
After the extraction, the remaining solid was dried under vacuum,
dissolved in a 1 M aqueous ammonia solution, and analyzed by UV−
vis spectroscopy.
(
(
3.84 mmol) based on the ammonium carbamate starting material
26 mmol), corresponding to 75 catalyst turnovers. The experiment
was reproducible. The turnovers were calculated by dividing the yield
of the urea by the catalyst loading.
Synthesis of Tetraammineaquacopper(II) Sulfate (Complex
70
1
). Complex 1 was synthesized using a literature method, where
Catalytic Urea Synthesis Using Complex 1, with 28.9 μL of
Water, and at 120 °C (Entry 4 in Table 1). Complex 1 in a 1%
catalyst loading (64 mg, 0.26 mmol), ammonium carbamate in 2.0 g
copper(II) sulfate pentahydrate and excess aqueous ammonia were
combined, followed by precipitation by alcohol at 0 °C. The product
was purified by washing with ethanol and pentane. The synthesis is
highly reproducible. A purchased sample from Strem Chemicals was
also used for catalysis. A PXRD spectrum of this complex, along with
(26 mmol), and water in 28.9 ± 3.2 μL were added to a high-pressure
metal reactor under air. The water was added via a syringe. The
details of the syringe and needle are described in the General section.
An oven was preheated to 120 °C. The oven door was briefly
opened, a high-pressure reactor was placed in the oven, and the oven
door was shut quickly. The temperature in the oven dropped to 110
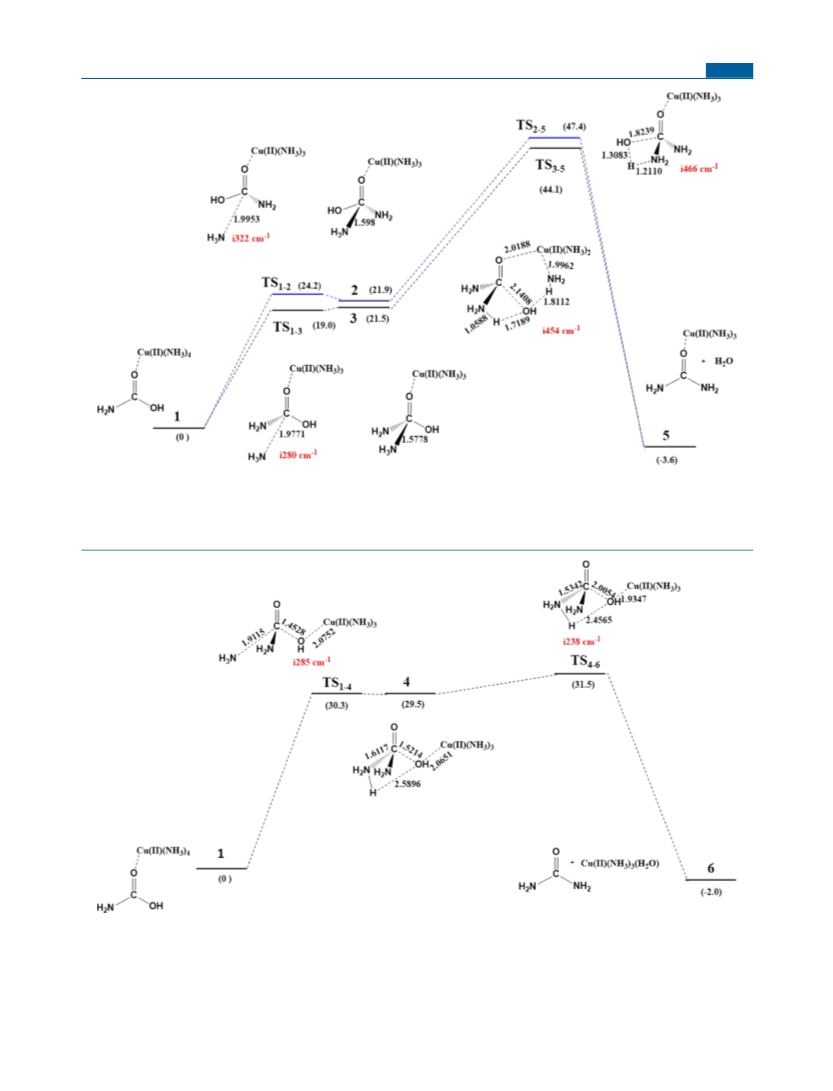
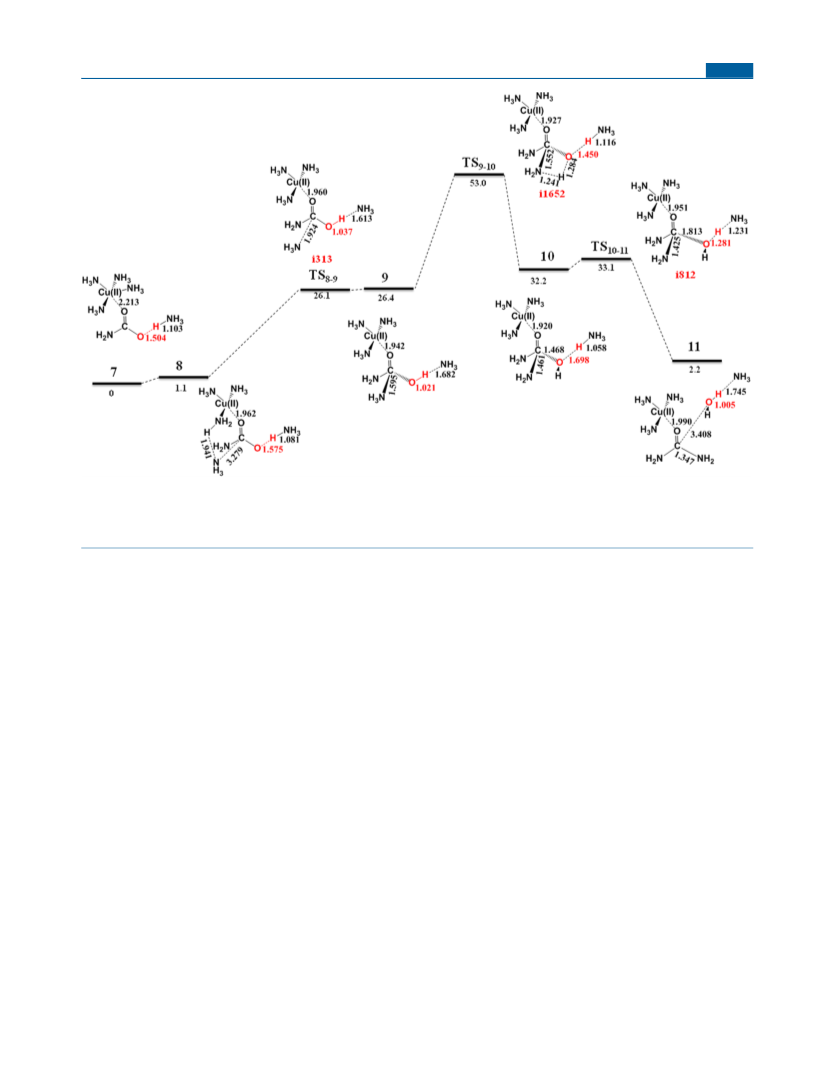
Calculation Methods. All structures are optimized at the B3LYP/
71,72
6
-31G** level of theory
with an SMD-water continuum solvent
°
C after placing the reactor. The heating lasted for 15 h, the oven was
turned off, and the door was opened to allow cooling by air. The
73
model, and the frequencies are calculated at the same level to
confirm that the structures are either minima (no imaginary
frequencies) or transition states (only one imaginary frequency).
The intrinsic reaction coordinate (IRC) method was employed to
follow the forward and reverse reaction paths for every transition
state, so that the corresponding reactants and products are
identified. All calculations are done with the Gaussian program
package. In section 3.2, the Gibbs free energies are additionally
estimated with single-point calculations at the M062x/6-311+
cooling took 30 min for the reactor to reach an ambient temperature
(
25 °C). Urea was detected at an 18 ± 6% yield, based on ammonium
1
13
carbamate, using a H{ C} NMR analysis in DMSO-d , where
6
benzene was an internal standard. The quantitative NMR analysis is
described in the Quantitative Analysis 1H{13C} NMR Spectroscopy
section.
74
UV−Vis Analysis of Copper(II) Biuret. At the end of the
reaction described in entry 1 of Table 1, the product mixture was
dissolved in water (5 mL). A 1 mL portion of this solution was diluted
to 2 mL and analyzed by UV−vis spectroscopy. No absorption for
75
+
G(3df,2pd) + SMD-water level of theory, which is denoted as
M062x/6-311++G(3df,2pd)//B3LYP/6-31G** + SMD-water. The
sign “//” means “using the optimized structures of”. The DFT
calculations with both B3LYP and M062x are done with the
unrestricted formula, UB3LYP or UM062X. The Mulliken spin
density on Cu(II) is ∼0.74 in all species, and the spin densities are
predominantly localized on Cu(II). The SMD solvent method was
specifically developed to predict free energies of solvation by using
35
copper biuret at 540 nm was detected.
1
13
Quantitative Analysis of Urea Using Solution-Phase H{ C}
NMR Spectroscopy. A Varian 400 MHz or an Anasazi 60 MHz
1
13
NMR spectrometer was used for the quantitative H{ C} analysis of
urea. The data processing was performed using ACD Spectrus
Processor software. At the end of the reaction described in the
5
586
Inorg. Chem. 2021, 60, 5573−5589

 Dennis, Donovan
Dennis, Donovan