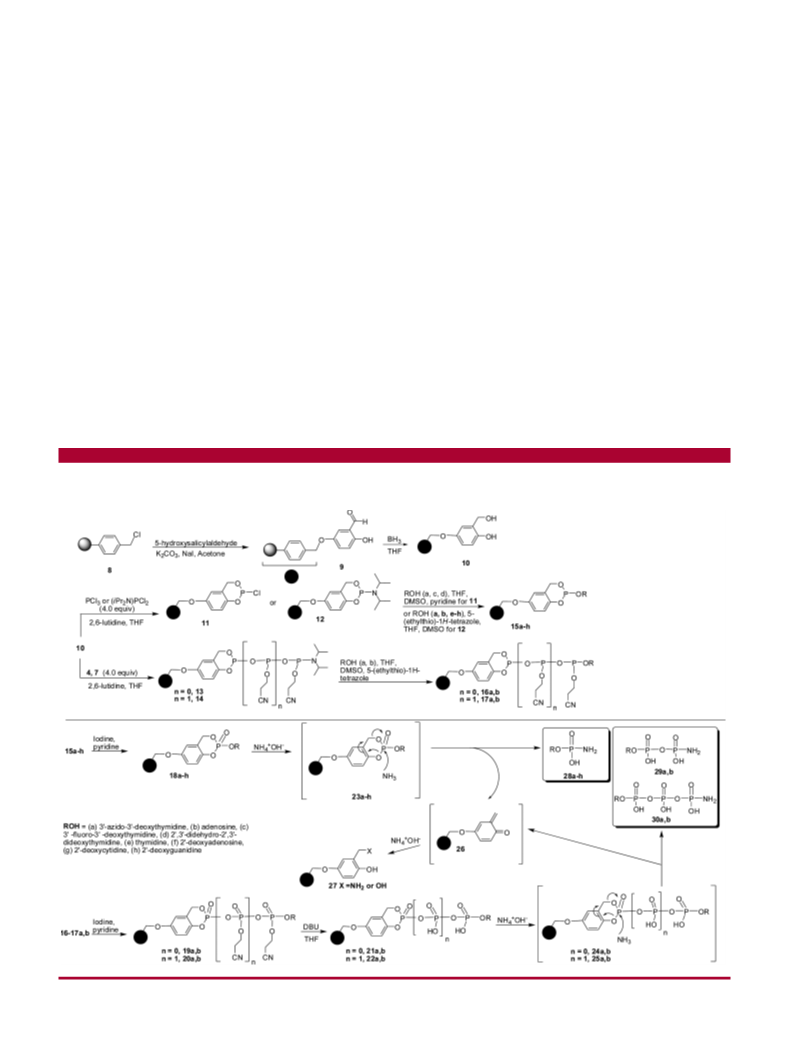
Scheme 1. Synthesis of Diphosphitylating and Triphosphitylating Reagents (4 and 7)
because of their stability toward nucleases and being able
to form a duplex with complementary chains of DNA or
cycloSaligenyl (cycloSal) phosphate triesters of several
nucleoside analogues have been designed as a pH-driven
9
,10
25-28
RNA sequences with higher affinity.
Catalysis of many
nucleotide delivery system.
As part of our ongoing
2
9
hydrolases and nucleases also occurs through nucleoside
efforts to synthesize organophosphorus compounds, we
report the synthesis of immobilized cycloSal phosphitylating
reagents and their application for the synthesis of nucleoside
mono-, di-, and triphosphoramidates to circumvent one or
more of the problems associated with the solution-phase
methods. To the best of our knowledge, this is the first paper
on the synthesis of polymer-bound cycloSal phosphitylating
reagents. Mono-, di-, and triphosphitylating reagents were
first immobilized on polystyrene resin-bound linker of
2-hydroxybenzyl alcohol. Coupling reactions of unprotected
nucleosides with the immobilized reagent followed by iodine
oxidation, deprotection, and basic cleavage afforded nucleo-
side mono-, di-, and triphosphoramidates.
11,12
phosphoramidate intermediates.
Therefore, the synthesis
of nucleoside phosphoramidates and phosphoramidate-based
pronucleotides and oligonucleotides is the subject of con-
siderable interest in nucleic acid research. The facile synthesis
of larger quantities of phosphoramidate derivatives is es-
sential for studying their biological properties.
The reported solution-phase methods for the synthesis of
nucleoside 5′-phosphoramidates include the reaction of nucleo-
side diphosphates, triphosphates, chlorophosphates, H-phos-
13-16
phonates, or trimethaphosphates with amines
in the
presence of a base and/or a coupling reagent (e.g., N-carbo-
1
3,17,18
15,19
diimide derivatives
Alternatively, highly reactive phosphoramidate precursors
e.g., phosphoryldichloride derivatives or bis(benzotriaz-
or trimethylsilyl chloride
).
The advantages of this solid-phase strategy included the
following: (i) The immobilization of hindered phosphitylating
reagents on a rigid polymer-bound linker allowed for the
regioselective reaction with the most reactive hydroxyl group
in the presence of an excess of unprotected nucleosides to
afford monosubstituted final products. (ii) Unprotected
nucleosides were used instead of precursor nucleoside
phosphate derivatives. (iii) Excesses of nucleosides and
unreacted reagents were removed in each step by washing
the resins. Furthermore, the modified linker remained trapped
on the resins. This facilitated isolation and purification of
monosubstituted final products. (iv) This strategy allowed
the synthesis of nucleoside 5′-O-mono-, di-, and triphos-
phoramidates from the same polymer-bound linker.
(
olyl)phosphoramidates) have been used in reaction with
nucleosides for the synthesis of nucleoside phosphorami-
6
dates. These methods have one or more disadvantages, such
as the requirement for the synthesis of precursor nucleoside
phosphates or phosphoramidates, the poor solubility of
precursors in organic solvents, tedious purification of final
products from intermediates and starting reagents, and low
or moderate overall yields. We have previously reported the
solid-phase synthesis of nucleoside mono-, di-, and triphos-
phates with high regioselectivity using polymer-bound linkers
20-24
of p-hydroxybenzyl alcohol or p-acetoxybenzyl alcohol.
(
9) Manoharan, M. Antisense Nucleic Acid Drug DeV. 2002, 12, 103
.
Scheme 1 illustrates the synthesis of diphosphitylating and
(10) Chen, J.-K.; Schultz, R. G.; Lioyd, D. H.; Gryaznov, S. M. Nucleic
Acids Res. 1995, 23, 2661
.
triphosphitylating reagents (4 and 7). Phosphorus trichloride
(
(
11) Huang, K.; Frey, P. A. J. Am. Chem. Soc. 2004, 126, 9548
.
12) Bieganowski, P.; Garrison, P. N.; Hodawadekar, S. C.; Faye, G.;
Barnes, L. D.; Brenner, C. J. Biol. Chem. 2002, 277, 10852
13) Parang, K.; Kohn, J. A.; Saldanha, S. A.; Cole, P. A. FEBS Lett.
002, 520, 156
.
(21) Parang, K.; Fournier, E. J.-L.; Hindsgaul, O. Org. Lett. 2001, 3,
(
307.
2
.
(22) Parang, K. Bioorg. Med. Chem. Lett. 2002, 12, 1863
(23) Ahmadibeni, Y.; Parang, K. J. Org. Chem. 2005, 70, 1100
(24) Ahmadibeni, Y.; Parang, K. Org. Lett. 2005, 7, 5589
(25) Meier, C. Mini-ReV. Med. Chem. 2002, 2, 219
(26) Meier, C.; Meerbach, A.; Balzarini, J. Front. Biosci. 2004, 9, 873
(27) Balzarini, J.; Aquaro, S.; Knispel, T.; Rampazzo, C.; Bianchi, V.;
Perno, C. F.; De Clercq, E.; Meier, C. Mol. Pharmacol. 2000, 58, 928
.
(
(
(
(
14) Zhu, J. G.; Fu, H.; Jiang, Y. Y.; Zhao, Y. F. Synlett 2005, 1927
15) Zhu, J.; Hua, F.; Jiang, Y.; Zhao, Y. J. Org. Chem. 2006, 71, 1722
16) Wray, J.; Jahn, W. FEBS Lett. 2002, 518, 97
17) Abraham, T. W.; Kalman, T. I.; McIntee, E. J.; Wagner, C. R.
.
.
.
.
.
.
.
J. Med. Chem. 1996, 39, 4569
18) Kruse, C. H.; Holden, K. G.; Offen, P. H.; Pritchard, M. L.; Field,
J. A.; Rieman, D. J.; Bender, P. E.; Ferguson, B.; Greig, R. G.; Poste, G.
J. Med. Chem. 1988, 31, 1768
19) Zhu, J.; Han, B.; Fu, H.; Jiang, Y.; Zhao, Z. J. Org. Chem. 2005,
0, 6676.
20) Ahmadibeni, Y.; Parang, K. Curr. Protoc. Nucleic Acid Chem. 2008,
Chapter 13, Unit 13.8.
.
(
.
(28) Jessen, H. J.; Fendrich, W.; Meier, C. Eur. J. Org. Chem. 2006,
.
974
.
(
(29) (a) Ahmadibeni, Y.; Parang, K. Org. Lett. 2005, 7, 1955. (b)
Ahmadibeni, Y.; Parang, K. J. Org. Chem. 2006, 71, 5837. (c) Ahmadibeni,
Y.; Parang, K. Angew. Chem., Int. Ed. 2007, 46, 4739. (d) Ahmadibeni,
Y.; Parang, K. Org. Lett. 2007, 9, 4483.
7
(
2158
Org. Lett., Vol. 11, No. 10, 2009

 Ahmadibeni, Yousef
Ahmadibeni, Yousef