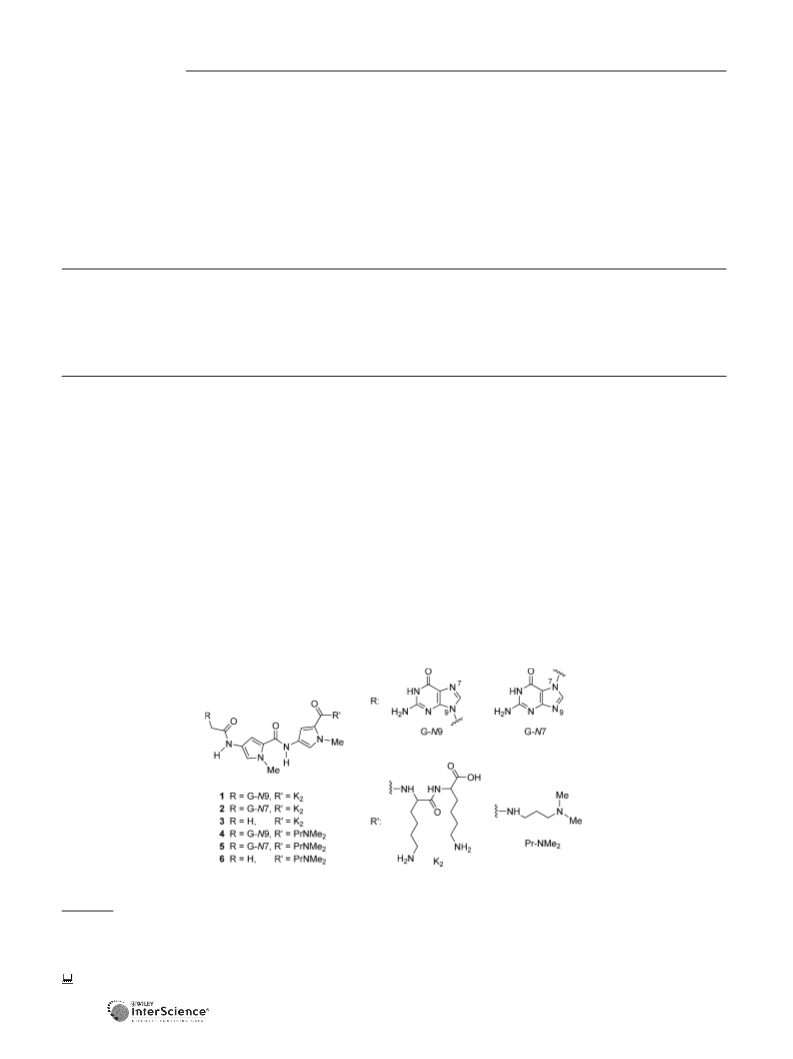
Guanine-Containing DNA Minor-Groove Binders
containing ligands were manually assembled in a polypropylene sy-
ringe fitted with a polyethylene filter disc. The following instru-
ments were used to obtain spectroscopic data: Nicolet 510 FT-IR,
Jasco V-550 with Peltier ETC-505T (UV and thermal denaturation
experiments), Jasco J-810 spectropolarimeter fitted with a thermo-
statted cell holder (CD experiments), Varian Mercury 400 MHz
(NMR), Bruker Digital Avance 600 MHz (NMR), HP-5988A (CI
1.41 (s, 9 H, Boc) ppm. 13C NMR ([D6]DMSO, 50 MHz): δ = 162.4
(COOH), 153.0 (C=O Boc), 123.4 (C-2), 120.2 (C-3), 119.3 (C-4),
108.0 (C-5), 79.0 (Cq Boc), 36.7 (NCH3), 28.8 (CH3 Boc) ppm. IR
(KBr): ν = 3351, 2979, 1686, 1586, 1451, 1246, 1111 cm–1. MS (ES,
˜
positive mode): m/z = 241.3 [M + H]+ (calcd. monoisotopic mass:
240.3).
Synthesis of Guaninylacetic Acids 15 and 16
MS), VG-Quattro (ES MS), Perseptive Biosystems Voyager DETM
RP with a 337 nm N2 laser (MALDI-TOF MS).
-
N2-Acetylguanine (12): Acetic anhydride (16.5 mL, 174.5 mmol)
was added to a suspension of guanine (11; 10 g, 66.2 mmol) in N,N-
dimethylacetamide, and the mixture was heated at 160 °C for 18 h.
The resulting solution was cooled to room temperature, and a pre-
cipitate was formed, which was filtered and washed with absolute
ethanol. The filtrate was concentrated under vacuum, which af-
forded more precipitate that was also filtered and washed. This
procedure was repeated until no precipitate was formed upon con-
centration of the solution. All the solid material was combined to
give 15.1 g of N2,N9-diacetylguanine [97% yield; MS (MALDI-
TOF, sinapinic acid, negative mode): m/z = 233.9 (calcd. monoiso-
topic mass: 235.1)]. N2,N9-Diacetylguanine (2.99 g, 12.7 mmol) was
suspended in ethanol/water (1:1, v/v; 15 mL), and the mixture was
heated at reflux for 2 h. Upon cooling, a precipitate appeared. The
mixture was concentrated to dryness, and the resulting solid was
co-evaporated with acetonitrile (3ϫ) to give 2.42 g of 12 (98%
yield). 1H NMR ([D6]DMSO, 400 MHz): δ = 7.96 (s, 1 H, 8-H),
2.14 (s, 3 H, N2-COCH3) ppm. MS (MALDI-TOF, sinapinic acid,
positive mode): m/z = 194.1 [M + H]+, 216.1 [M + Na]+, 232.0 [M
+ K]+ (calcd. monoisotopic mass: 193.1).
Synthesis of 4-Boc-amino-N-methylpyrrole-2-carboxylic Acid (10)
Methyl N-Methyl-4-nitropyrrole-2-carboxylate (8): Treatment of N-
methylpyrrole (7) with trichloroacetyl chloride followed by reaction
with nitric acid and then sodium methoxide to afford compound 8
was carried out as described in ref.[14] Rf (hexanes/ethyl acetate, 1:1,
v/v) = 0.6. 1H NMR ([D6]DMSO, 200 MHz): δ = 8.26 (d, J =
1.8 Hz, 1 H, 5-H), 7.23 (d, J = 1.8 Hz, 1 H, 3-H), 3.89 (s, 3 H,
NCH3), 3.77 (s, 3 H, OCH3) ppm. 13C NMR ([D6]DMSO,
50 MHz): δ = 159.8 (CO), 134 (C-5), 129.5 (C-4), 122.6 (C-2), 111.5
(C-3), 51.9 (OCH ), 37.6 (NCH ) ppm. IR (KBr): ν = 3149, 1717,
˜
3
3
1542, 1420, 1318, 1195, 1115, 752 cm–1. MS (CI, NH3): m/z = 202
[M + NH4]+ (calcd. monoisotopic mass: 184.0).
Reduction of the Nitro Group and Isolation of 9: Compound 8
(500 mg, 1.72 mmol) was introduced into a round-bottomed flask
and dissolved in a 6:1 mixture of anhydrous methanol/dichloro-
methane (12 mL). 10% Pd/C (165 mg) and ammonium formate
(788 mg) were subsequently added, and the mixture was purged
with Ar. The flask was gently heated to promote initiation of the
reaction, which was identified by the formation of CO2 bubbles.
After 2.5 min, trifluoroacetic acid (1.25 mL, 16.8 mmol) was
added. The reaction mixture was filtered to remove the solid cata-
lyst. Elimination of the solvent under reduced pressure afforded
2.44 g of crude 9 mixed with salts, which was used without purifica-
tion. MS (ES, positive mode): m/z = 155.2 [M + H]+ (calcd. mono-
isotopic mass: 154.2).
Methyl (N2-Acetylguanin-x-yl)acetate (x = 9: 13; x = 7: 14): DIEA
(4.3 mL, 25.2 mmol) and methyl bromoacetate (1.3 mL,
13.7 mmol) were added to a suspension of 12 (2.42 g, 12.5 mmol)
in DMF (36 mL) under Ar. After 20 h of stirring at room tempera-
ture, the solvent was removed in vacuo, and the residue was co-
evaporated with methanol (3ϫ). The resulting crude product was
resuspended in methanol and added slowly and with vigorous stir-
ring to water (95 mL) in an Erlenmeyer flask. The mixture was
filtered under vacuum to afford 14 as a solid (1.35 g, 41% yield).
The filtrate was concentrated under vacuum and chilled to yield a
second solid product (13), which was also isolated by filtration un-
der vacuum (0.96 g, 29% yield).
Protection of the Amine and Ester Hydrolysis: Introduction of the
Boc group was carried out essentially as described previously,[14]
but by using 3 rather than 1.1 equiv. of di-tert-butyl dicarbonate.
The resulting N-Boc-protected ester was characterized by NMR
1
and MS. H NMR ([D6]DMSO, 200 MHz): δ = 9.10 (s, 1 H, NH),
7.08 (s, 1 H, 5-H), 6.59 (s, 1 H, 3-H) 3.76 (s, 3 H, NCH3), 3.68 (s,
3 H, OCH3), 1.34–1.41 (s, 9 H, Boc + small impurity of tert-butyl
alcohol and Boc2O) ppm. 13C NMR ([D6]DMSO, 50 MHz): δ =
161.4 (CO carboxylate), 153.4 (C=O Boc), 123.8 (C-2), 119.9 (C-
4), 119.3 (C-3), 108.0 (C-5), 79.2 (Cq Boc), 51.5 (OCH3), 36.7
(NCH3), 28.8 (CH3 Boc) ppm. MS (CI, NH3): m/z = 255
[M + H]+, 272 [M + NH4]+ (calcd. monoisotopic mass: 254.1).
Finally, the N-Boc-protected pyrrole ester (1.42 g) was dissolved in
methanol/water (3:1, v/v; 20 mL), and LiOH was added (1.17 g,
27.9 mmol). The mixture was heated at 45 °C, and the progress of
the reaction was monitored by TLC (hexanes/ethyl acetate, 3:1, v/
v). When the starting material could no longer be detected, meth-
anol was eliminated under reduced pressure. The aqueous solution
was poured into water/ethyl acetate (1:1, v/v; 30 mL), and H2SO4/
water (1:1, v/v) was added with vigorous stirring, until the pH of
the aqueous phase was 2–2.5. The two layers were separated, and
the organic phase was dried with MgSO4. Removal of the solvent
under reduced pressure afforded 542 mg of pure 10 (8 Ǟ 10 yield:
83%).
1
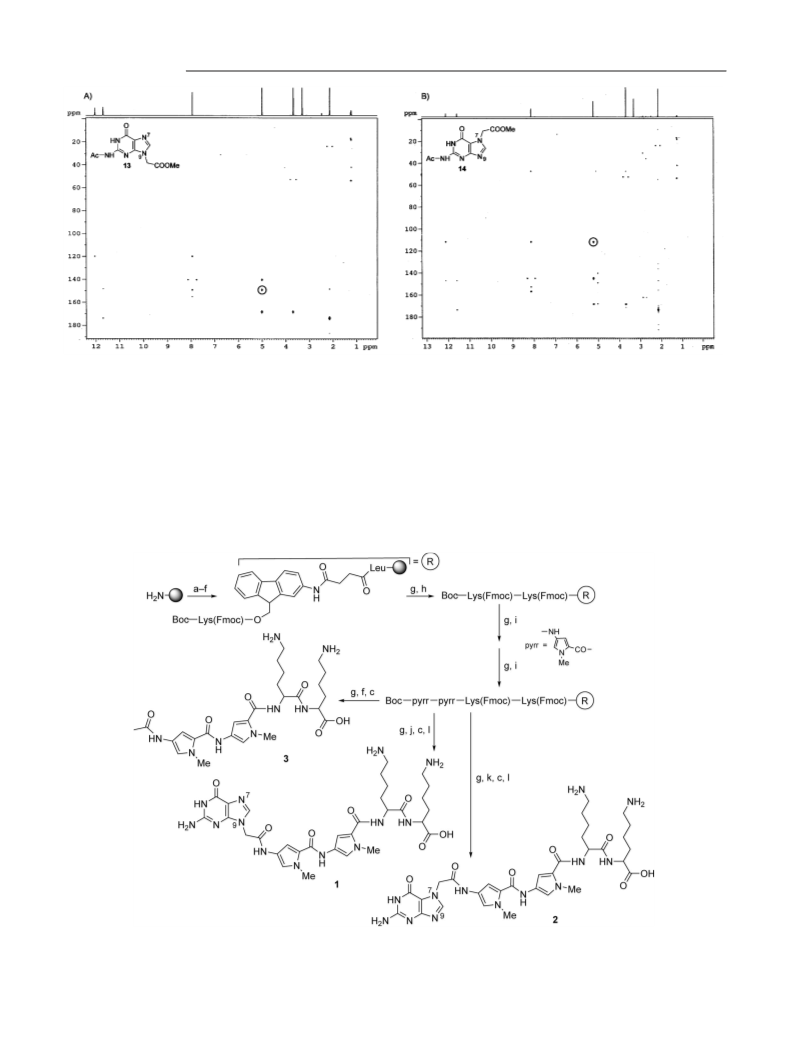
Methyl (N2-Acetylguanin-9-yl)acetate (13): H NMR ([D6]DMSO,
400 MHz): δ = 12.05 (s, 1 H, N1-H), 11.69 (s, 1 H, N2-H), 7.95 (s,
1 H, 8-H), 5.01 (s, 2 H, CH2COO), 3.69 (s, 3 H, COOCH3), 2.15 (s,
3 H, COCH3) ppm. 13C NMR ([D6]DMSO, 100 MHz): δ = 174.2
(COCH3), 168.8 (COO), 155.5 (C-2), 149.7 (C-4), 148.7 (C-6), 140.9
(C-8), 120.4 (C-5), 53.2 (COOCH3), 44.9 (CH2COO), 24.4
(NHCOCH3) ppm. [1H-13C] HMBC ([D6]DMSO, 600 MHz): see
Figure 2a. MS (MALDI-TOF, sinapinic acid, positive mode): m/z
= 266.1 [M + H]+, 288.1 [M + Na]+, 304.1 [M + K]+ (calcd. mono-
isotopic mass: 265.1).
1
Methyl (N2-Acetylguanin-7-yl)acetate (14): H NMR ([D6]DMSO,
400 MHz): δ = 12.10 (s, 1 H, N1-H), 11.60 (s, 1 H, N2-H), 8.13 (s,
1 H, 8-H), 5.21 (s, 2 H, CH2COO), 3.69 (s, 3 H, COOCH3), 2.15
(s, 3 H, NHCOCH3) ppm. 13C NMR ([D6]DMSO, 100 MHz): δ =
174.1 (COCH3), 169.1 (COO), 157.6 (C-4), 153.3 (C-2), 147.8 (C-
6), 145.6 (C-8), 112.4 (C-5), 53.2 (COOCH3), 47.9 (CH2COO), 24.4
(NHCOCH3) ppm. [1H-13C] HMBC ([D6]DMSO, 600 MHz): see
Figure 2b. MS (MALDI-TOF, sinapinic acid, positive mode): m/z
= 266.2 [M + H]+, 288.2 [M + Na]+, 304.1 [M + K]+ (calcd. mono-
isotopic mass: 265.1).
Characterization of 10: Rf (hexanes/ethyl acetate, 3:1, v/v) = 0.4; Rf
(AcOEt) = 0.81. M.p. 157–160 °C (ref.[16] 160–161 °C). 1H NMR
([D6]DMSO, 200 MHz): δ = 12.1 (s, 1 H, COOH), 9.05 (s, 1 H, (Guanin-x-yl)acetic Acid (x = 9: 15; x = 7: 16): Compound 13
NH), 7.02 (s, 1 H, 5-H), 6.55 (s, 1 H, 3-H), 3.75 (s, 3 H, NCH3), (905 mg, 3.41 mmol) or 14 (900 mg, 3.39 mmol) was suspended in
Eur. J. Org. Chem. 2009, 1398–1406
© 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
www.eurjoc.org
1403

 Pulido, Daniel
Pulido, Daniel