Full Paper
Synthesis of PY3: Following the procedure outlined for the synthe- Synthesis of PH2: Following the procedure outlined for the synthe-
sis of PY1, 7c was treated with triphenylphosphine. Compound PY3
was obtained as a red solid in 44 % yield. M.p. ≈102–104 °C. 1H
NMR (400 MHz, CDCl3): δ = 7.93–7.74 (m, 12 H, -ArH), 7.73–7.62 (m,
7 H, -ArH), 7.50 (d, J = 8.4 Hz, 2 H, -ArH), 7.08 (d, J = 9.6 Hz, 1 H,
-ArH), 7.03 (d, J = 3.7 Hz, 1 H, -ArH), 6.96 (d, J = 8.4 Hz, 2 H, -ArH),
6.67 (d, J = 8.2 Hz, 2 H, -ArH), 3.99 (t, J = 5.8 Hz, 2 H, -OCH2), 3.86
(t, J = 6.6 Hz, 2 H, -CH2), 3.38 [q, J = 6.9 Hz, 4 H, -N(CH2)2], 1.81–
1.73 (m, 4 H, -2CH2), 1.63–1.69 (m, 2 H, -CH2), 1.63–1.69 (m, 2 H,
-CH2), 1.18 (t, J = 6.9 Hz, 6 H, -2CH3) ppm. 13C NMR (101 MHz,
CDCl3): δ = 160.69 (-C-OCH2), 157.15 (-C=O), 147.49 (-ArC), 144.58
sis of PY4, 13 was treated with morpholine and potassium carb-
onate. Compound PH2 was obtained as an orange-red oil in 88 %
yield. H NMR (400 MHz, CDCl3): δ = 8.56 (d, J = 7.5 Hz, 1 H, -ArH),
7.82–7.72 (m, 2 H, -ArH), 7.66–7.72 (m, 2 H, -ArH), 7.58 (d, J = 8.5 Hz,
2 H, -ArH), 7.48 (d, J = 8.8 Hz, 2 H, -ArH), 6.98–7.06 (m, 3 H, -ArH),
6.65 (d, J = 8.8 Hz, 2 H, -ArH), 4.04 (t, J = 6.2 Hz, 2 H, -OCH2), 3.73
(t, J = 4.5 Hz, 4 H, -2CH2), 3.35 [q, J = 7 Hz, 4 H, -N(CH2CH3)2], 2.53–
2.45 (m, 4 H, -2CH2), 2.43 (t, J = 7.5 Hz, 2 H, -CH2), 1.90–1.80 (m, 2
H, -CH2), 1.76–1.65 (m, 2 H, -CH2), 1.16 (t, J = 7 Hz, 6 H, -2CH3) ppm.
13C NMR (101 MHz, CDCl3): δ = 160.05 (-C-OCH2), 156.43 (-C=O),
1
(-ArC), 141.84 (-ArC), 140.71 (-ArC), 135.08 (d, J = 3.0 Hz, -PPh3), 147.66 (-ArC), 147.16 (-ArC), 140.55 (-ArC), 140.43 (-ArC), 132.86
133.85 (d, J = 10.1 Hz, -PPh3), 130.60 (-ArC), 130.59 (d, J = 12.6 Hz, (-ArC), 131.71 (-ArC), 131.01 (-ArC), 128.81 (-ArC), 128.32 (-ArC),
-PPh3), 128.85 (-ArC), 128.72 (-ArC), 127.79 (-ArC), 127.01 (-ArC), 128.16 (-ArC), 127.57 (-ArC), 127.14 (-ArC), 126.74 (-ArC), 121.73
126.72 (-ArC), 119.35 (-ArC), 118.92 (-ArC), 118.61 (d, J = 86.2 Hz, (-ArC), 118.46 (-ArC), 118.20 (-ArC), 114.59 (-ArC), 111.87 (-ArC),
-PPh3), 115.16, 111.97, 68.04 (-OCH2), 44.54 (-CH2CH3), 30.11 [d, J = 67.83 (-OCH2), 66.92 (-OCH2CH2N), 58.59 (-NCH2CH2O), 53.70
15.8 Hz, -CH2(CH2)2PPh3], 29.81 (-CH2PPh3), 28.88 (-OCH2CH2), 25.74
[-O(CH2)2CH2], 22.79 (d, J = 3.06 Hz, -CH2CH2PPh3), 12.68 (-CH2CH3)
[-NCH2(CH2)3O], 44.40 (-CH2CH3), 27.16 [-N(CH2)2CH2CH2O], 23.05
[-NCH2CH2(CH2)2O], 12.67 (-CH2CH3) ppm. HRMS (ESI): calcd. for
ppm. HRMS (ESI): calcd. for C48H49N3O2PS 762.3278 [M]+; found C36H41N4O3S 609.2899 [M + H]+; found 609.2879.
762.3206.
Determination of the Fluorescence Quantum Yield of the PY
and PH Derivatives: The quantum yields of one-photon emission
of the synthesized PY and PH derivatives were measured with rhod-
amine B (RhB, Φ = 0.72, dissolved in MeOH) as a reference. The
one-photon fluorescence measurements were performed in 1 cm
Synthesis of PY4: In
a round-bottomed flask, 7b (80 mg,
0.14 mmol), morpholine (38 mg, 0.43 mmol), and potassium carb-
onate (60 mg, 0.43 mmol) were mixed in acetonitrile (10 mL), and
the mixture was heated at refluxed for 10 h. After cooling, the mix-
ture was filtered, and the filtrate was concentrated under reduced
pressure. The residue was purified by chromatography (silica gel,
dichloromethane/methanol 20:1) to afford PY4 (68 mg, 85 %) as a
quartz cells with 1 μM compound in DMSO or CH2Cl2 with a fluores-
cence lifetime and steady-state spectrophotometer (Edinburgh In-
strument FLS920) equipped with a 450 W Xenon light and 2.5 × 2.5
slits. The values of fluorescence quantum yield, Φ (sample), were
calculated according to the following equation:[15]
1
red solid. M.p. ≈114–116 °C. H NMR (400 MHz, CDCl3): δ = 7.83 (d,
J = 6.1 Hz, 2 H, -ArH), 7.81 (s, 1 H, -ArH), 7.65 (d, J = 9.7 Hz, 1 H,
-ArH), 7.51 (d, J = 8.7 Hz, 2 H, -ArH), 7.11 (d, J = 9.6 Hz, 1 H, -ArH),
7.05 (d, J = 4.1 Hz, 1 H, -ArH), 6.99 (d, J = 8.7 Hz, 2 H, -ArH), 6.69 (d,
J = 8.8 Hz, 2 H, -ArH), 4.05 (t, J = 6.1 Hz, 2 H, -OCH2), 3.77 (t, J =
6.1 Hz, 4 H, -2CH2), 3.39 [q, J = 7 Hz, 4 H, -N(CH2CH3)2], 2.62–2.42
(m, 6 H, -3CH2), 1.91–1.82 (m, 2 H, -CH2), 1.80–1.67 (m, 2 H, -CH2),
in which Φref is the value of the fluorescence quantum yield of the
reference (ΦRhB = 0.72),[16] I is the integrated emission intensity, OD
1.19 (t, J = 7 Hz, 6 H, -2CH3) ppm. 13C NMR (101 MHz, CDCl3): δ = is the optical density at the excitation wavelength, and d is the
160.60 (-C-OCH2), 157.15 (-C=O), 147.49 (-ArC), 144.47 (-ArC), 141.89 refractive index of the solvent (dDMSO = 1.478, d
= 1.444, dH O =
CH2Cl2
2
(-ArC), 140.68 (-ArC), 130.65 (-ArC), 128.60 (-ArC), 127.81 (-ArC),
127.04 (-ArC), 126.94 (-ArC), 121.56 (-ArC), 119.40 (-ArC), 118.95
(-ArC), 115.08 (-ArC), 111.95 (-ArC), 67.92 (-OCH2), 66.82
(-OCH2CH2N), 58.66 (-NCH2CH2O), 53.71 [-NCH2(CH2)3O], 44.56
(-CH2CH3), 27.20 [-N(CH2)2CH2CH2O], 22.98 [-NCH2CH2(CH2)2O],
12.76 (-CH2CH3) ppm. HRMS (ESI): calcd. for C32H39N4O3S 559.2743
[M + H]+; found 559.2692.
1.333, dMeOH = 1.329).
Cell Culture: All cells were incubated in complete medium [Dul-
becco's modified Eagle's Medium, supplemented with 10 % fetal
bovine serum (FBS) and 1 % penicillin–streptomycin] at 37 °C under
an atmosphere containing 5 % CO2. For imaging, RD cells were
grown in poly-
plete medium (2 mL) for 24 h. Cells were washed with phosphate-
buffered saline (PBS), and stocked dyes (2 m in DMSO) were added
to obtain a final concentration of 2 μ . The treated cells were incu-
D-lysine-coated dishes and were incubated in com-
M
Synthesis of PH1: Following the procedure outlined for the synthe-
sis of PY4, 13 was treated with triphenylphosphine. Compound PH1
was obtained as a red solid in 78 % yield. M.p. ≈115–117 °C. 1H
NMR (400 MHz, CDCl3): δ = 8.57 (d, J = 7.7 Hz, 1 H, -ArH), 7.87–7.73
(m, 12 H, -ArH), 7.710–7.63 (m, 7 H, -ArH), 7.57 (d, J = 8.5 Hz, 2 H,
-ArH), 7.46 (d, J = 7.9 Hz, 2 H, -ArH), 7.00 (m, 3 H, -ArH), 6.65 (d, J =
7.6 Hz, 2 H, -ArH), 4.20 (t, J = 5.7 Hz, 2 H, -OCH2), 4.03–3.91 (m, 2
H, -CH2), 3.36 [q, J = 6.7 Hz, 4 H, -N(CH2)2], 2.31–2.20 (m, 2 H, -CH2),
1.94–1.85 (m, 2 H, -CH2), 1.16 (t, J = 6.9 Hz, 6 H, -2CH3) ppm. 13C
M
bated for 30 min in the dark at 37 °C. A few minutes prior to confo-
cal imaging, cells were washed with PBS (2×). A confocal laser scan-
ning microscope (A1R-si, Nikon, Japan) was used to obtain images.
Cells were imaged by the fluorescence mode with a 40× immersion
lens with the following parameters: laser power 3 %; pinhole 1.0 au;
excitation wavelength 405, 488, 561, or 633 nm; resolution 512 ×
512; and a scan speed of 0.5 frames s–1
.
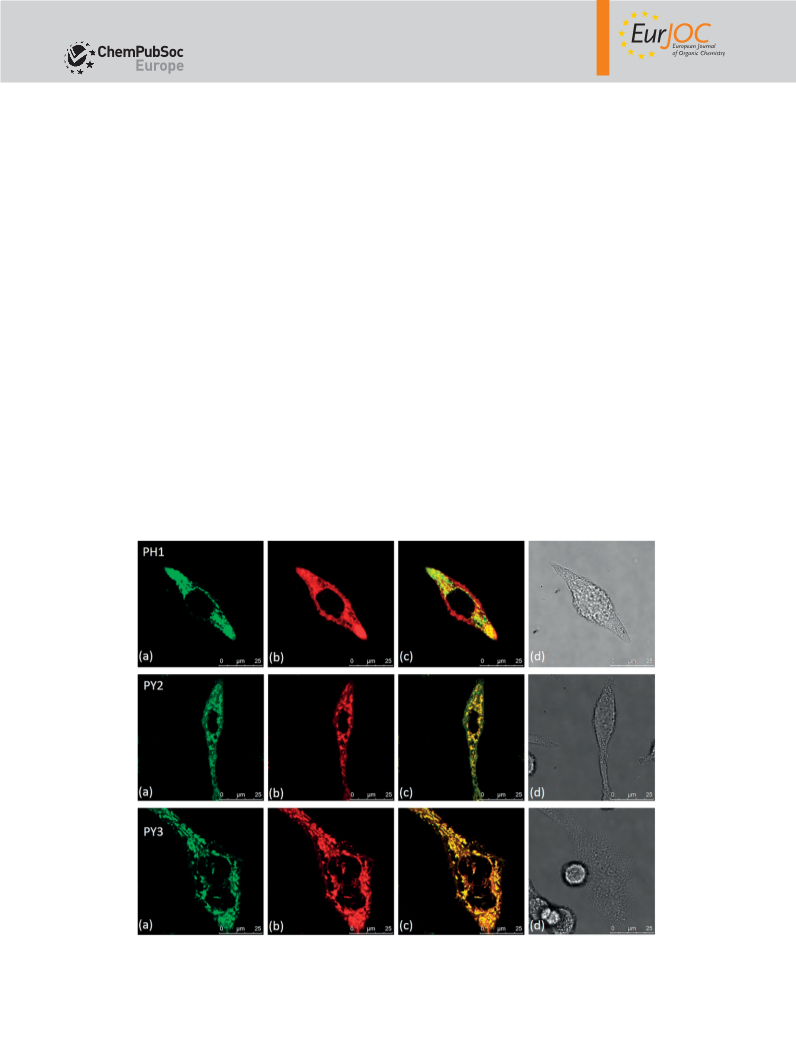
NMR (101 MHz, CDCl3): δ = 159.85 (-C-OCH2), 156.59 (-C=O), 147.78 Colocalization Assay: RD cells were placed onto 0.1 m
M
poly-
lysine-coated glasses in complete medium, and the cells were incu-
bated for 24 h. A stock solution of PY or PH in chromatographic
grade, anhydrous DMSO was prepared as 2 m . The solution was
diluted to a final concentration of 2 μ by adding complete growth
medium. Stock solutions of Mito-Tracker Green FM, Lyso-Tracker
Green DND-26 were prepared as 1 m , and the stock solution was
diluted to the working concentration in complete medium (100 n ).
After incubating for 30 min, cells were washed with PBS buffer (2×)
D-
(-ArC), 147.22 (-ArC), 140.95 (-ArC), 140.66 (-ArC), 135.10 (d, J =
2.9 Hz, -PPh3), 133.79 (d, J = 9.9 Hz, -PPh3), 133.09 (-ArC), 131.89
(-ArC), 131.11 (-ArC), 130.96 (-ArC), 130.57 (d, J = 12.4 Hz, -PPh3),
128.86 (-ArC), 128.38 (-ArC), 128.21 (-ArC), 127.66 (-ArC), 127.27
(-ArC), 127.17 (-ArC), 126.82 (-ArC), 118.38 (d, J = 85.9 Hz, -PPh3),
118.29 (-ArC), 114.77 (-ArC), 111.92 (-ArC), 66.78 (-OCH2), 44.45
(-CH2CH3), 29.34 (d, J = 16.8 Hz, -CH2PPh3), 22.15 (d, J = 50.7 Hz,
-OCH2CH2), 19.41 (d, J = 3.6 Hz, -CH2CH2PPh3), 12.69 (-CH2CH3) ppm.
M
M
M
M
HRMS (ESI): calcd. for C50H47N3O2PS 784.3121 [M]+; found 784.3059. before the confocal experiments. Images were taken under condi-
Eur. J. Org. Chem. 2017, 3274–3281
3280 © 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

 Zhou, Tongliang
Zhou, Tongliang