2
120
K. W. Temburnikar et al. / Bioorg. Med. Chem. 22 (2014) 2113–2122
1
NMR (400 MHz, DMSO-d
6
): d 8.22 (s, 1H), 11.40 (br s, 1H, NH),
1.58 (br s, 1H, NH). 13C NMR (100 MHz, DMSO-d
): d 99.6,
12.1, 133.4, 145.1, 152.1, 159.0. FAB-MS m/z for C BrN S cal-
Mp 224.6–227.9 °C. H NMR (400 MHz, CDCl
3
): d 8.23 (s, 1H), 8.29
): d 109.1, 116.5,
139.4, 143.2, 150.5, 154.1, 156.2, 162.8. FAB-MS m/z for C
13
1
1
6
(s, 1H), 9.42 (s, 1H). C NMR (100 MHz, CDCl
3
6
H
3
2
O
2
8
81
H
3-
+
79
81
+
35
culated [M+H] 246.9171, found 246.9175 ( Br), 248.9161 ( Br).
BrClN
3
5
S calculated [M+H] 315.9053, found 315.9059 ( Cl, Br),
17.9036 ( Cl, Br), 319.9009 ( Cl, Br).
3
5
81
37
81
5
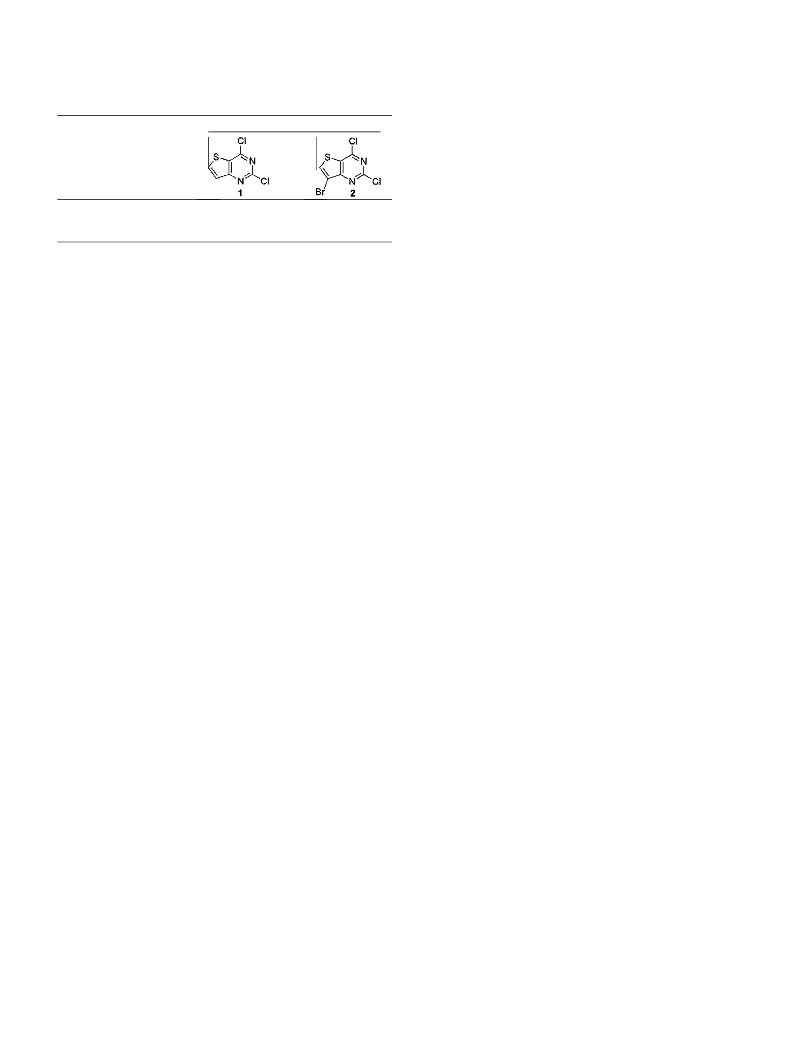
.1.1.7. 7-Bromo-2,4-dichlorothieno[3,2-d]pyrimidine (2).
a round bottom flask containing 7-bromothieno[3,2-d]pyrimidin-
,4-dione 8 (4.07 g, 16.47 mmol), DMAP (8.38 g, 68.76 mmol) and
freshly distilled POCl were added and the suspension stirred at
05–110 °C for 2 h under nitrogen. The POCl was evaporated
and the residue extracted with CH Cl (300 mL). The organic layer
was washed with aq NaHCO (300 mL), brine (200 mL) and dried
over MgSO . The dried organic layer was concentrated, loaded on
To
0
0
5.1.1.11. 2,4-Dimethoxy-7-(b-D-glycero-pentofuran-3 -ulos-1 -
yl)thieno[3,2-d]pyrimidine (13). In a dry flask 7-bromo-
2
3
2,4-dimethoxythieno[3,2-d]pyrimidine 9 (1.10 g, 4.01 mmol) was
co-evaporated with CH3CN (10 mL) and dried under high vacuum.
To this tetra-n-butyl ammonium chloride (7.81 g, 28.1 mmol) was
added and the mixture dissolved in DMF (10 mL). To this solution
47.5% Pd(OAc)2 (378 mg, 0.8 mmol) was added followed by NaH-
CO3 (674 mg, 8.02 mmol) under nitrogen. In a separate flask, glycal
(2.13 g, 6.01 mmol) was co-evaporated with CH3CN (10 mL) and
dissolved in DMF (10 mL). A solution of glycal in DMF was added
to the previous solution and the mixture stirred at 40 °C for 48 h
under nitrogen until the TLC indicated the absence of starting
materials. The reaction mixture was evaporated, the residue dis-
solved in CH2Cl2 (50 mL) and the Pd filtered over celite. The organ-
ic layer was loaded on silica and the product purified using column
chromatography eluting with 3:1 hexanes/EtOAc to afford 13 as a
1
3
2
2
3
4
silica and the product purified using column chromatography elut-
ing with 19:1 hexanes:EtOAc to obtain 2 as a white solid (3.41 g,
1
1
f
2.00 mmol, 73%). R 0.5 in 9:1 hexanes/EtOAc. Mp 180.2–
83.0 °C. H NMR (400 MHz, CDCl
1
13
3
): d 8.13 (s, 1H). C NMR
(
100 MHz, CDCl
3
): d 160.0, 158.0, 156.0, 135.0, 128.0, 109.5. FAB-
+
6 2 2
MS m/z for C HBrCl N S calculated [M+H] 282.8493, found
3
5
79
35
81
35
2
82.8495 (2x Cl, Br), 284.8468 (2x Cl, Br), 286.8443 ( Cl,
3
7
81
37
81
Cl, Br), 288.8419 (2x Cl, Br).
5
a
.1.1.8. 7-Bromo-2,4-dimethoxythieno[3,2-d]pyrimidine (9).
solution of 7-bromo-2,4-dichlorothieno[3,2-d]pyrimidine
To
2
pale yellow solid (650 mg, 2.09 mmol, 52%). Mp 123.2–126.9 °C. R
0.4 in 1:1 hexanes/EtOAc. The spectral data was in agreement with
f
2
3
(
(
3.6 g, 12.6 mmol) in anhydrous MeOH, 30% NaOMe solution
10 mL, 2.90 g, 54.5 mmol) was added and the reaction mixture
literature values. 1H NMR (400 MHz, CDCl3): d 2.80 (dd, 1H,
J = 6.4 Hz, J = 17.6 Hz), 3.23 (dd, 1H, J = 11.0 Hz, J = 17.6 Hz), 3.96–
3.98 (br m, 2H), 4.03 (s, 3H), 4.10 (t, 1H, J = 2.3 Hz), 4.16 (s, 3H),
4.89 (br d, 1H, OH), 5.50 (dd, 1H, J = 6.4 Hz, J = 11.0 Hz), 7.80 (s,
1H). 13C NMR (100 MHz, CDCl3): d 49.3, 54.5, 55.4, 62.7, 74.0,
82.3, 113.3, 133.4, 133.9, 160.3, 164.0, 166.1, 213.8. FAB-MS m/z
was refluxed overnight (16 h). TLC indicated the absence of start-
ing material upon which the reaction mixture was neutralized
(
pH 7–8) using 2 M HCl followed by removal of MeOH in vacuo.
The product was extracted with CH Cl (300 mL) and the organic
layer washed with water (300 mL) and brine (300 mL). The CH Cl
2
2
+
2
2
for C13H14N2O5S calculated [M+H] 311.0696, found 311.0695.
was evaporated completely and to the residue EtOAc was added to
obtain slurry that was heated to reflux and cooled to get a white
precipitate. The precipitate was filtered to obtain 9 as a white gran-
0
0
5.1.1.12.
1 -b-[7-(2,4-Dimethoxythieno[3,2-d]pyrimidine)]-2 -
deoxyribofuranose
(14).
2,4-Dimethoxy-7-(b-D-glycero-
0
0
ular solid (2.40 g, 8.72 mmol, 69%). R
f
0.5 in 9:1 hexanes/EtOAc. Mp
pentofuran-3 -ulos-1 -yl)thieno[3,2-d]pyrimidine 13 (650 mg,
1
data.
93–195 °C. The spectral data was in agreement with the reported
2.09 mmol) was dissolved in a mixture of CH
(5 mL) under nitrogen. The solution was cooled to ꢀ10 to ꢀ5 °C
after which NaB(OAc) H (555 mg, 2.62 mmol) was added and stir-
red at ꢀ10 to ꢀ5 °C for 1 h. The reaction mixture warmed to room
temperature and stirred until the TLC indicated the absence of the
starting material. The reaction mixture was concentrated, loaded
on silica and purified using column chromatography eluting with
3
CN (5 mL) and AcOH
2
3 1
1
H NMR (400 MHz, CDCl
3
): d 4.10 (s, 3H), 4.14 (s, 3H), 7.76
): d 54.9, 55.4, 108.7, 111.4, 131.2,
BrN S calculated
3
(
s, 1H). C NMR (100 MHz, CDCl
3
3
1
59.8, 164.9, 166.2. FAB-MS m/z for C
8
H
7
2
O
2
+
79
81
[
M+H] 274.9484, found 274.9490 ( Br), 276.9473 ( Br).
5
.1.1.9.
10).
d]pyrimidine 2 (100 mg, 0.35 mmol) was suspended in MeOH
4-Amino-7-bromo-2-chlorothieno[3,2-d]pyrimidine
(
In glass tube 7-bromo-2,4-dichloro thieno[3,2-
a
49:1 CH
72%). Mp 129.7–132.4 °C. R
(400 MHz, CDCl ): d 2.011 (dd, 1H, J = 5.3 Hz, J = 13.0 Hz), 2.76–
2
Cl
2
/MeOH to afford 14 as yellow solid (470 mg, 1.50 mmol,
/MeOH. 1H NMR
f
0.4 in 19:1 CH Cl
2
2
(
10 mL) and cooled to ꢀ60 to ꢀ70 °C at which point ammonia
3
was bubbled in the suspension. The glass tube was sealed and
the suspension stirred at room temperature overnight (16 h) to ob-
tain a clear solution. The solvent was evaporated and the resulting
crude compound was purified using column chromatography elut-
ing first with 9:1 hexanes/EtOAc, followed by 8:2 hexanes/EtOAc to
2.84 (m, 1H), 3.80 (dd, 1H, J = 1.4 Hz, J = 12.4 Hz), 3.95 (dd, 1H,
J = 2.1 Hz, J = 12.1 Hz), 4.06 (s, 3H), 4.18–4.15 (m, 1H), 4.16 (s,
3H), 4.72 (d, 1H, J = 5.0 Hz), 5.46 (dd, 1H, J = 5.5 Hz, J = 11.4 Hz),
1
3
7.73 (s, 1H). C NMR (100 MHz, CDCl
3
) 41.9, 54.3, 55.3, 64.0,
75.2, 88.9, 113.2, 133.0, 135.1, 160.6, 163.7, 166.0. FAB-MS m/z
+
obtain 10 as an off-white solid (65 mg, 0.24 mmol, 68%). R
3
d 8.97 (br s, 2H, NH
d 108.6, 114.0, 133.5, 158.6, 158.8, 160.8. FAB-MS m/z for
f
0.3 in
for C13
H
16
N
2 5
O S calculated [M+H] 313.0853, found 313.0862.
1
:1 hexanes/EtOAc. Mp 286–287.9 °C. H NMR (400 MHz, CDCl
3
):
):
), 9.14 (s, 1H). 13C NMR (100 MHz, CDCl
5.1.1.13. 1 -b-[7-(2,4-Dimethoxythieno[3,2-d]pyrimidine)]-3 ,5 -
0
0
0
2
3
0
0
acetoxy-2 -deoxyribofuranose (15).
(2,4-dimethoxythieno[3,2-d]pyrimidine)]-2 -deoxyribofuranose 14
To a solution of 1 -b-[7-
+
0
C
6
H
3
BrClN
3
S
calculated [M+H] 263.8992, found 263.9001
3
5
Cl, Br), 265.8974 ( Cl, 81Br), 269.8948 ( Cl, Br).
79
35
37
81
(
(470 mg, 1.50 mmol) in pyridine (5 mL), Ac
2
O
(0.28 mL,
3
.01 mmol) was added and stirred overnight (16 h) at which point
5
.1.1.10.
7-Bromo-2-chloro-4-[1,2,4-triazolo]thieno[3,2-d]
To solution of 7-bromo-2,4-dichlorothie-
no[3,2-d]pyrimidine 2 (100 mg, 0.35 mmol) in CH CN, 1,2,4-tria-
zole (73 mg, 1.05 mmol), Et N (0.18 mL, 1.26 mmol) were added
the TLC indicated the absence of starting material. The pyridine
was evaporated and the residue co-evaporated with toluene. The
residue was then loaded on silica and the product purified using
column chromatography eluting with 19:1 then 9:1 hexanes/
EtOAc to give product 15 as a pale yellow solid (570 mg,
pyrimidine (11).
3
3
and stirred overnight at which point a thick white precipitate
was obtained. The reaction mixture was loaded onto silica and
the product purified using column chromatography eluting with
1.43 mmol, 95%). Mp 79.6–83.2 °C. R
f
0.2 in 3:1 hexanes/ EtOAc.
1
H NMR (400 MHz, CDCl
3
): d 2.03 (s, 3H), 2.12 (s, 3H), 2.15–2.23
1
9:1, then 9:1 and finally 4:1 hexanes/EtOAc to obtain 11 as a
(m, 1H), 2.64 (ddd, 1H, J = 1.4 Hz, J = 5.5 Hz, J = 13.7 Hz), 4.04 (s,
3H), 4.12 (s, 3H), 4.23–4.26 (m, 2H), 4.42 (dd, 1H, J = 3.2,
white solid (77 mg, 0.24 mmol, 69%). R 0.2 in 19:1 hexanes/EtOAc.
f

 Temburnikar, Kartik W.
Temburnikar, Kartik W.