M. Bartlam, L.-L. Wong et al.
than propane, NADPH consumption assays were carried out by
using 0.1–0.25 mm enzyme and otherwise as described.[24] Propyl-
benzene, toluene, fluorene and 3-methylpentane turnovers were
analysed as described.[23,24] The products from fatty acid turnovers
were derivatised[47] and analysed as described[23] except that for
palmitic acid, the GC injector temperature was maintained at
2508C and the flame-ionisation detector at 3008C. The products
from palmitic acid oxidation were assumed to be as reported,[37]
the GC retention times for the corresponding derivatives being
11.6 min (13-hydroxypalmitic acid), 12.0 min (14-hydroxypalmitic
acid) and 12.2 min (15-hydroxypalmitic acid). KM and kcat values
were derived by fitting initial NADPH consumption rates as a func-
tion of substrate concentration to a rectangular hyperbola by
using Origin 8 software (Origin Labs). Kinetic titrations with palmit-
ic acid were run in phosphate (50 mm, pH 7.4) at 30ꢁ0.18C, and
the volume of DMSO was held constant at 1% (v/v). Substrate con-
centrations ranged from 5 to 200 mm, and enzyme concentrations
were fixed at 0.1 mm.
rates so far reported for P450BM3 with any substrate. With tolu-
ene, R3IP gave the highest PFR (1824 vs. 2.1 minꢀ1 for WT).
This was because coupling was more efficient when the A330P
mutation was present (about 50% for R3 and R3IP vs. about
15% for RLYFIP and I401P).
Variant RLYFIP gave a PFR of 2980 minꢀ1 with 3-methylpen-
tane versus 28 minꢀ1 for WT, more than twice that given by
WT with lauric acid. The NADPH consumption rate and cou-
pling efficiency were both significantly enhanced. 3-Methyl-
pentan-2-ol was the major product with all the variants investi-
gated (75–88%). With propane, R3IP gave a PFR of 430 versus
0.2 minꢀ1 for WT. As with toluene, the ability of A330P-contain-
ing variants to utilise NADPH efficiently with small substrates
was influential, R3IP and R3 coupling at 29 and 21%, respec-
tively, against 8.5% for RLYFIP and 0.7% for WT. Propan-2-ol
was the dominant product with all variants, though WT and
A330P-containing variants also formed small quantities of
propan-1-ol. The oxidation of propane by a custom-created 24-
mutation P450BM3 variant has been reported, the kcat value at
Propane turnovers were carried out in 4000 mL volumes with
0.25 mm enzyme. Propane was bubbled into Tris (50 mm, pH 7.4,
2% (v/v) DMSO) on ice for 30 min while oxygen was bubbled into
a separate reservoir of Tris (50 mm, pH 7.4), also on ice. P450BM3
and bovine liver catalase (400 mg) were added to an appropriate
volume of oxygen-saturated buffer, followed by propane-saturated
buffer (3000 mL). The full cuvette was promptly sealed, inverted
several times, and held at 30ꢁ0.18C for 2 min prior to NADPH ad-
dition through the silicone seal to a final concentration equivalent
to 1 AU from a 40 mgmLꢀ1 stock in Tris (50 mm, pH 7.4). Product
analysis was carried out as for 3-methylpentane,[23] by using
propan-2-ol as calibrant and pentan-3-ol as internal standard. The
SPB-1 column temperature was raised from 60 to 808C at
28Cminꢀ1, then at 708Cminꢀ1 to 2208C. The product retention
times were 3.73 min (propan-2-ol) and 4.83 min (propan-1-ol).
258C being 455 minꢀ1 [44]
.
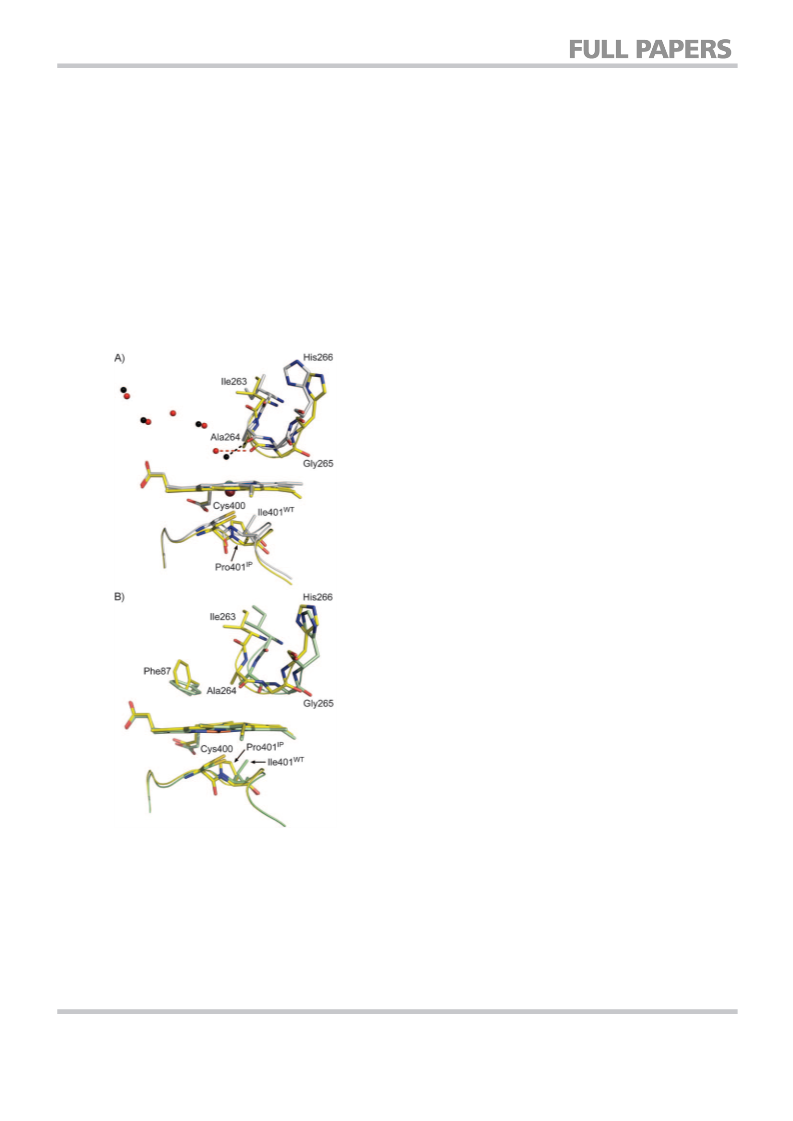
In conclusion, the two crystal structures reported in this
paper give contrasting perspectives on how the substitution of
proline into an enzyme can alter catalytic properties. In A330P,
dramatic but localised changes impact the active site. The
I401P mutation causes more complex and extensive structural
perturbation, the trigger for which may be the displacement of
the haem iron away from the substrate pocket. Proline is un-
suitable for introduction at many of the sites usually targeted
for mutagenesis in P450BM3 as it is conformationally demand-
ing. However, where the structure is able to accommodate
such an aggressive substitution, unexpected and desirable out-
comes can result.
Reduction potential measurements: Redox titrations were carried
out for the haem domains of WT and variants by varying the
applied potential and fitting the resulting spectra to the sums of
spectra from the oxidised and reduced forms of the protein by
using the Nernst equation, as described.[23] The spectrum of the re-
duced form was confirmed by treatment of the oxidised form with
a slight excess of dithionite. Because the reduced form of methyl
viologen, one of the mediators, absorbed strongly in the Soret
region, spectra were fitted to the absorbances at 465 nm, a wave-
length reported as being suitable for analysis,[48] and also 450 and
570 nm. Fitting the 415–700 nm region of the spectrum gave iden-
tical midpoint potentials, within experimental error. The data
shown in Figure S3 in the Supporting Information were fitted at
465 nm. All potentials relate to the standard hydrogen electrode;
those given for SF WT and SF I401P differ from values published
previously (by ꢂ1%) as additional data have been acquired.
Experimental Section
DNA manipulations, protein expression, purification and quanti-
tation: Preparation of the haem domains of A330P and I401P was
as described.[23] Variants R47L/Y51F/I401P (RLYFIP) and R47L/Y51F/
A330P/I401P (R3IP) were prepared by standard cloning proce-
dures[45] and site-directed mutagenesis was performed by using
the Stratagene QuikChange mutagenesis kit. Expression and purifi-
cation of haem domains and full-length proteins for in vitro use
was carried out as described.[23,24] Because I401P variants were par-
tially high-spin in the resting state,[23] determination of the A418/A280
absorbance ratio did not represent an appropriate method of
purity assessment. Instead, the A404/A280 absorbance ratio was as-
sessed, 404 nm being the isosbestic point. This typically fell in the
0.3–0.4 range for full-length proteins and 0.7–0.8 for haem do-
mains. Protein concentration was determined from reduced CO-
bound spectra.[46]
Stopped-flow spectrophotometry: The rate constant for the for-
mation of the FeII(CO) complex, kf, by the full-length enzymes was
determined at 25.0ꢁ0.18C by monitoring the absorbance at
450 nm by using an Applied Photophysics SX20 stopped-flow
spectrophotometer in single wavelength mode. Experiments were
carried out in a Belle Technology glove box ([O2]ꢂ5 ppm). Compo-
nents were prepared in Tris (50 mm, pH 7.4, 2% (v/v) DMSO) satu-
rated with CO by gently bubbling for 5 min and then purging the
headspace for a further 5 min. One syringe contained NADPH
(200 mm), and the other the appropriate P450BM3 variant. In SB
enzyme measurements, the second syringe also contained palmitic
acid (200 mm). The components were mixed in a 1:1 ratio. Enzyme
In vitro oxidation: Assays were run on a Varian Cary 50 spectro-
photometer. All data were means of at least three experiments.
Standard deviations, where not explicitly stated, are ꢂ5% of the
mean. Experiments involving fatty acids were carried out by using
DMSO solutions of the acid itself rather than the corresponding
salt. Palmitic acid (ꢂ300 mm) was added from a 10 mm stock. In all
cases, NADPH consumption rates were derived by using e340
=
6.22 mmꢀ1 cmꢀ1 and are stated without adjustment for the rate re-
corded in the absence of substrate (leak rate). For substrates other
2554
ꢀ 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
ChemBioChem 2010, 11, 2549 – 2556

 Whitehouse, Christopher J. C.
Whitehouse, Christopher J. C.