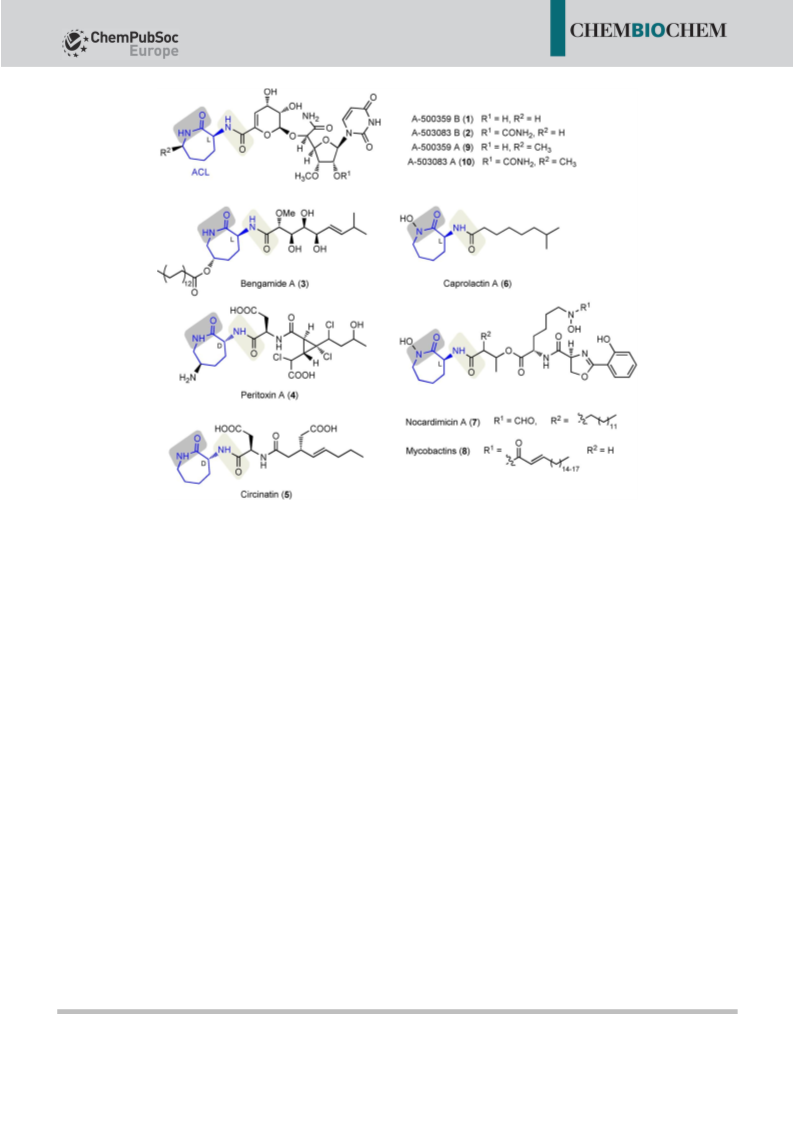
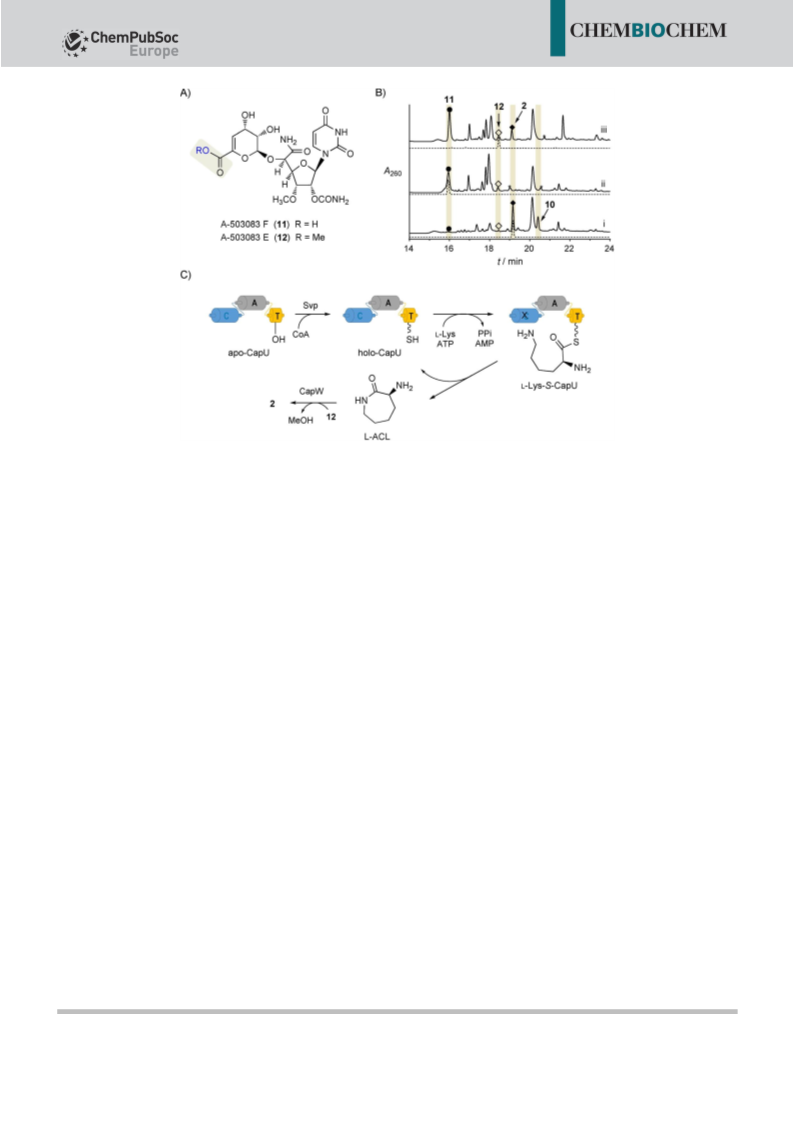
Communications
After sequencing to confirm PCR fidelity, pUWL201pw-capUV was
transformed into S. lividans TK64 by PEG-mediated protoplast
transformation.[34] After 20 h at 288C, plates were overlaid with soft
nutrient agar (1 mL) supplemented with thiostrepton (200 mg).
After three additional days at 288C, single colonies were trans-
ferred to fresh R2YE plates supplemented with thiostrepton
(50 mgmLÀ1). After 4 d at 288C, positive transformants were con-
firmed by colony PCR with InstaGene Matrix from Bio-Rad (Her-
cules, CA) and LA-Taq polymerase with GC buffer II from Takara Bio
(Shiga, Japan). Positive transformants were used to inoculate R2YE
(50 mL) containing thiostrepton (25 mgmLÀ1) and grown for 2 d at
288C at 250 rpm, at which point 2 mL was transferred to fresh
R2YE (100 mL) containing thiostrepton (25 mgmLÀ1). After growth
for 3 d at 288C and at 250 rpm, l-Lys was added (25 mgmLÀ1 final
concentration), and the culture was grown for an additional 72 h.
The culture was separated by centrifugation, and the pH of the su-
pernatant was adjusted to 7 with NaOH (1m). The mixture was ap-
plied to a column containing XAD16 resin and sequentially washed
with 5 volumes each of water, acetone (20%), acetone (50%), and
acetone (80%). Fractions eluting with acetone (20%) were collect-
ed and dried under vacuum. The dried solid containing l-ACL from
a 100 mL culture was dissolved in sodium bicarbonate solution
(500 mL, 20 mm), and an equal volume of Fmoc-OSu acetonitrile
solution (2 mm) was added. After the mixture had been stirred for
5 min, samples were analyzed with an Agilent 1200 Series Quater-
nary LC system equipped with an Eclipse XDB-C18 column
(150 mm4.6 mm, 5 mm, 80 ). A series of linear gradients was de-
veloped from TFA (0.1%) in water (A) to TFA (0.1%) in acetonitrile
(90%, B) in the following manner (beginning time and ending time
with linear increase to % B): 0–20 min, 50–90% B; 20–25 min, 90%
B. The flow rate was kept constant at 1.0 mLminÀ1, and elution
was monitored at 254 nm.
One-pot reaction with CapU, CapV and CapW: Reaction mixtures
(100 mL) consisted of Tris·Cl (pH 7.5, 50 mm), l-Lys (5 mm), CoA
(0.05 mm), ATP (5 mm), MgCl2 (20 mm), 12 (2 mm), Svp (3.5 mm),
CapU (3.5 mm), CapV (3.5 mm), and CapW (3.5 mm), at 308C for 8 h.
After removal of protein by ultrafiltration, the reaction components
were analyzed by reversed-phase chromatography with use of the
Dionex Ultimate 3000 system equipped with an Acclaim 120 C-18
column (4.6 mm100 mm, 3 mm). A series of linear gradients from
TFA (0.1%) in acetonitrile (2.5%, C) to TFA (0.1%) in acetonitrile
(90%, D) was developed in the following manner (start time and
end time with linear increase to % D): 0–5 min 0% D, 5–10 min 0–
50% D, 10–15 min 50–100% D, and 15–18 min 100% D. The flow
rate was kept constant at 1 mLminÀ1, and elution was monitored
at 260 nm. Peaks were identified by retention time and MS by
comparison with control reactions or authentic standards.
Formation and analysis of l-ACL: Reaction mixtures (100 mL) con-
sisted of Tris·HCl or phosphate (50 mm, pH 7.5), l-Lys (5 mm), ATP
(5 mm), MgCl2 (20 mm), Svp (3.5 mm), CapU/truncated CapU (A+
T domains, 3.5 mm), and CapV or CapW (3.5 mm), at 308C for 2 h.
After removal of the protein by ultrafiltration, the reaction compo-
nents (50 mL) were treated with sodium bicarbonate (1m, 50 mL)
and dansyl chloride (25 mL of 10 mgmLÀ1 in DMF). After incubation
for 1 h at 428C, reaction components were analyzed by use of the
Agilent system described above. A series of linear gradients from
formic acid (0.1%) in water (E) to formic acid (0.1%) in acetonitrile
(F) was developed in the following manner (start time and end
time with linear increase or decrease of % F): 0–18 min 5–90% F
and 18–20 min 90–5% F. The flow rate was kept constant at
0.4 mLminÀ1, and elution was monitored at 310 nm. Peaks were
identified by comparison with the control reaction and authentic
standards.
Synthesis of l-Lys-SNAC (13): Diisopropylethylamine (1.4 mL,
8.0 mmol) and N-acetylcysteamine (0.22 mL, 2.2 mmol) were se-
quentially added to a solution of N-Boc-l-Lys (0.692 g, 2.0 mmol)
and PyBOP (2.0 g, 4.0 mmol) in CH2Cl2 (10 mL). The mixture was
stirred at room temperature, and the reaction was monitored by
TLC until completion (ꢀ2 h). The solvent was removed under
vacuum, and the residue was dissolved in ethyl acetate (50 mL).
The organic layer was washed with brine (210 mL) and dried to
give a colorless oil. N-Boc-l-Lys-SNAC was purified by silica column
chromatography (isocratic EtOAc/hexane 1:1) as a white solid. N-
Boc-l-Lys-SNAC was dissolved in CH2Cl2 (2 mL) at room tempera-
ture, and TFA (2 mL) was added dropwise. The resulting mixture
was stirred for 1 h at room temperature, and the solvent was
removed under vacuum. Compound 13 was purified by silica
column chromatography as a colorless solid in 62% yield over two
Cloning and expression of capU, capV, and capU_AT: The genes
for CapU, CapV, and CapU_AT (truncated CapU consisting of the A
and T domains) were amplified by PCR with use of an Expand
Long Template PCR System from Roche with supplied buffer 2,
dNTPs (200 mm), DMSO (5%), pN-4 (10 ng),[12] DNA polymerase
(5 U), and the primer pairs (400 nm each) capU (forward) 5’-GGTAT
TGAGG GTCGC ATGGA CGCCC CGCGT CACTG-3’/(reverse) 5’-
AGAGG AGAGT TAGAG CCTCA GGGCG AGGAG TCGAC ATAG-3’,
capV (forward) 5’-GGTAT TGAGG GTCGC ATGCC CGGAC CGCAG
AATG-3’/(reverse) 5’-AGAGG AGAGT TAGAG CCTCA CCAAC GGAAT
GTCCC G-3’, and capU_AT (forward) 5’-AAAAA ACATA TGGGT
GATGT CATCG GCCCC-3’/(reverse) 5’-AAAAA AGGAT CCTCA GGGCG
AGGAG TCGAC A-3’. The PCR program included an initial hold at
948C for 2 min, followed by 30 cycles of 948C for 10 s, 568C for
15 s, and 688C for 60 s. The gel-purified PCR product of capU and
capV was inserted into pET-30 Xa/LIC by ligation-independent clon-
ing as described by Novagen (Madison, WI, USA) to yield pET30-
capU and pET30-capV, which were subsequently sequenced to con-
firm PCR fidelity. The gel-purified PCR product of capU_AT was di-
gested with NdeI and BamHI and inserted into similarly digested
Pdb·His·MBP by using T4 DNA ligase (New England Biolabs) to
yield Pdb·His·MBP-capU_AT, which was sequenced to confirm PCR
fidelity. The capV gene, engineered to be expressed as an N-termi-
nal His6-protein, was subcloned by PCR with pET30-capV as the
template and the primer pair (forward) 5’-GATAG GCATA TGCCC
GGACC GCAGA ATG-3’ (NdeI site underlined)/(reverse) 5’-CGAGT
TAAGC TTTCC CCAAC GGAAT GTCCC GG-3’ (HindIII site underlined).
After restriction digestion the PCR product was ligated into a
pET30 DNA fragment originating from pET30-capV that was digest-
ed with the same enzymes. The resulting plasmid, pET30-CcapV,
was used for production of the C-terminal His6-tagged CapV.
1
steps. H NMR (400 MHz, [D4]methanol): d=4.18 (t, J=6.3 Hz, 1H),
3.32–3.43 (m, 2H), 3.03–3.19 (m, 2H), 2.93 (t, J=7.6 Hz, 2H), 1.91–
2.06 (m, 2H), 1.90 (s, 3H), 1.65–1.75 (m, 2H), 1.43–1.58 ppm (m,
2H); 13C NMR (100 MHz, D2O with methanol as standard): d=
197.92, 173.65, 60.33, 49.99, 40.29, 39.71, 32.27, 29.82, 28.07,
22.76 ppm; HRMS (ESI): m/z calcd for C10H21N3O2S+H+:248.1427;
found: 248.1424.
Synthesis of l-Lys-PANT (14): 1) 2,2-Dimethoxypropane (4.88 mL,
40 mmol) was added to a solution of d-pantethine (1.526 g,
2 mmol) and PTSA (0.038 g, 0.2 mmol) in acetone (10 mL). The mix-
ture was stirred at room temperature for 8 h, at which point the
solvent was removed under vacuum. Isopropylidene-d-pantethine
was purified by silica column chromatography (5–10% methanol/
ethyl acetate mixture) as a white powder. 2) Isopropylidene-d-pan-
tethine (0.635 g, 1.0 mmol) was dissolved in sodium bicarbonate
solution (1m, 5 mL) containing dithiothreitol (0.185 g, 1.2 mmol),
ChemBioChem 2016, 17, 804 – 810
809
ꢀ 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

 Liu, Xiaodong
Liu, Xiaodong