Arkivoc 2019, v, 0-0
Mujahid, M. et al.
Conclusions
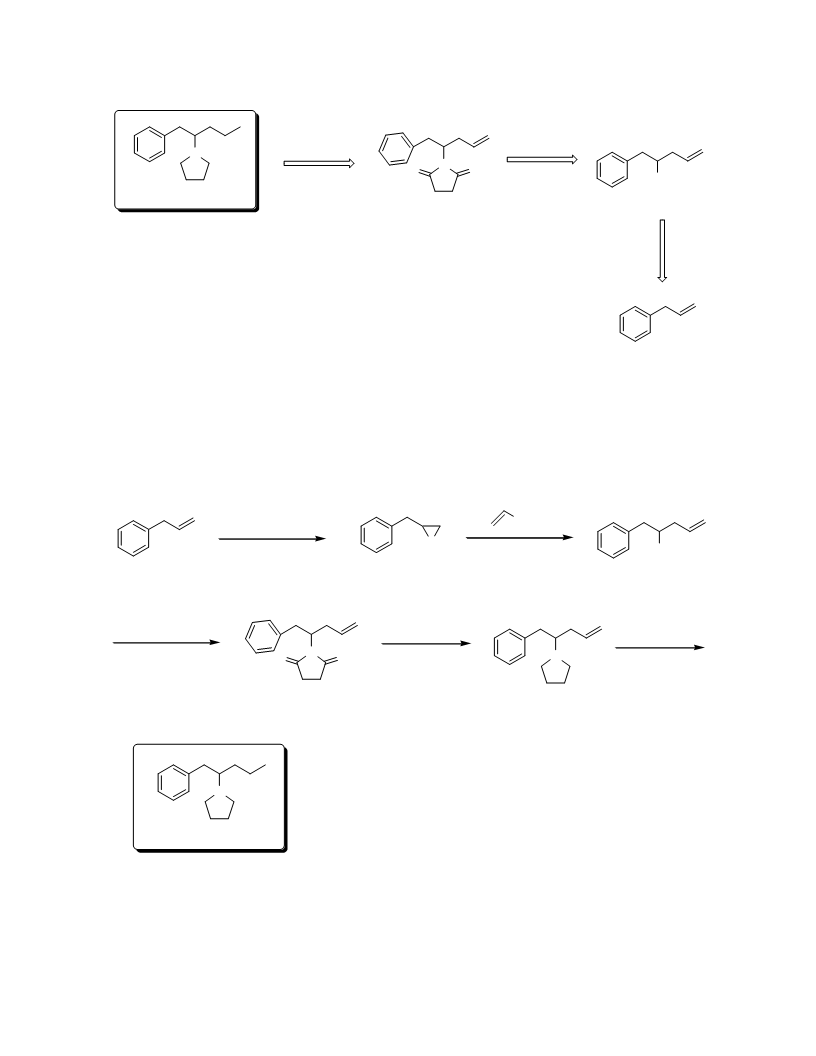
In summary, we have demonstrated a new method for the preparation of central nervous system (CNS)
stimulant prolintane 1. Commercially available starting material, simple transformations and good overall yield
are some of the salient features of this approach. Using this protocol, chiral analogues of the targeted compound
can be prepared by employing hydrolytic kinetic resolution (HKR) strategy on intermediate 3. We envisage that
the present protocol may be useful for the synthesis of other prolintane analogues which may be required for
extensive biological studies.
Experimental Section
General. Solvents were purified and dried by standard procedures prior to use. IR spectra were obtained from
Perkin–Elmer Spectrum One spectrophotometer. 1H NMR and 13C NMR spectra were recorded on a Bruker AC-
200 NMR spectrometer. Spectra were obtained in CDCl3. Monitoring of reactions was carried out using TLC
plates Merck Silica Gel 60 F254 and visualization with UV light (254 and 365 nm), I2 and anisaldehyde in ethanol
as development reagents.
2-Benzyloxirane (3). To a solution of allylbenzene (2) (5.9 mL, 45.0 mmol) in DCM (200 mL) was added, 3-
chloroperoxy benzoic acid (m-CPBA) (11.6 g, 67.5 mmol) slowly portionwise under vigorous stirring at 0°C within
30 min. The reaction mixture was allowed to attain room temperature and stirred for 12 h. After completion of
the reaction, the resultant solution filtered under reduced pressure and neutralized by 200 mL sodium hydrogen
carbonate solution (5.6 g, 67.5 mmol). The organic layer separated and washed with water (3 × 50 mL) then
dried over anhydrous magnesium sulfate. Solvent was removed under reduced pressure and the crude mixture
was purified over column chromatography (silica gel, petroleum ether/EtOAc, 96:4) to afford 3 as a colorless oil
(4.1 g, 68%); 1H NMR (200 MHz, CDCl3): δH 7.26-7.37 (m, 5H), 3.09-3.19 (m, 1H), 2.89-3.00 (m, 1H), 2.82-2.87 (m,
1H), 2.76-2.79 (m, 1H), 2.56 (dd, J 2.7, 5.1 Hz, 1H); l3C NMR (50 MHz, CDCl3): δC 136.8, 128.9, 128.3, 126.3, 51.9,
46.5, 38.5; Anal. Calcd for C9H10O (134.18): C, 80.56; H, 7.51. Found C, 80.21; H, 7.43 %.
1-Phenylpent-4-en-2-ol (4). To a pre cooled (-20 oC) solution of epoxide 3 (4.0 g, 29.8 mmol) and CuI (0.1 g) in
dry THF (40 mL) was added vinyl magnesium chloride (1.0 M in THF, 40 mL, 32.8 mmol) in THF for about 30 min.
Subsequently, the reaction mixture was allowed to attain ambient temperature and continued the stirring for
additional 1 h. After completion of the reaction (indicated by TLC), aqueous NH4Cl (20 mL) was added, after
which the reaction mixture was filtered, and washed with ethyl acetate. The solvent was removed under
reduced pressure and the crude product was subjected to column chromatography (silica gel, petroleum
1
ether/ethyl acetate, 90:10) to yield compound 4 as a colorless oil (4.4 g; 85%); H NMR (200 MHz, CDCl3): δH
7.31 - 7.39 (m, 2H), 7.20-7.30 (m, 3H), 5.87-5.92 (m, 1H), 5.14-5.24 (m, 2H), 3.90-3.93 (m, 1H), 2.86 (dd, J 13.4,
4.9 Hz, 1H), 2.76 (dd, J 13.4, 7.9 Hz, 1H), 2.32-2.43 (m, 1H), 2.21-2.32 (m, 1H), 1.87 (br. s, 1H): l3C NMR (50 MHz,
CDCl3): δC 138.4, 134.7, 129.4, 128.5, 126.4, 118.0, 71.6, 43.2, 41.1; Anal. Calcd for C11H14O (162.23): C, 81.44;
H, 8.70. Found C, 81.21; H, 8.52 %.
1-(1-Benzylbut-3-enyl)pyrrolidine-2,5-dione (5). A solution of DIAD (5.3 mL, 26.6 mmol) in dry THF (5 mL) was
added dropwise to a solution of compound 4 (3.9 g, 22.2 mmol), succinimide (2.64 g, 26.6 mmol) and
triphenylphosphine (6.9 g, 26.6 mmol) in dry THF (60 mL) under N2 atmosphere at 0 °C. The reaction mixture
was stirred at ambient temperature for 5 h. The solvent was removed under reduced pressure and the residue
was purified by column chromatography (silica gel, petroleum ether/ethyl acetate, 85:15) to yield compound 5
Page 4
©ARKAT USA, Inc

 Mujahid
Mujahid