
Journal of Medicinal Chemistry p. 6658 - 6661 (2004)
Update date:2022-08-11
Topics:
 Lombardo, Louis J.
Lombardo, Louis J.
Lee, Francis Y.
Chen, Ping
Norris, Derek
Barrish, Joel C.
Behnia, Kamelia
Castaneda, Stephen
Cornelius, Lyndon A. M.
Das, Jagabandhu
Doweyko, Arthur M.
Fairchild, Craig
Hunt, John T.
Inigo, Ivan
Johnston, Kathy
Kamath, Amrita
Kan, David
Klei, Herbert
Marathe, Punit
Pang, Suhong
Peterson, Russell
Pitt, Sidney
Schieven, Gary L.
Schmidt, Robert J.
Tokarski, John
Wen, Mei-Li
Wityak, John
Borzilleri, Robert M.
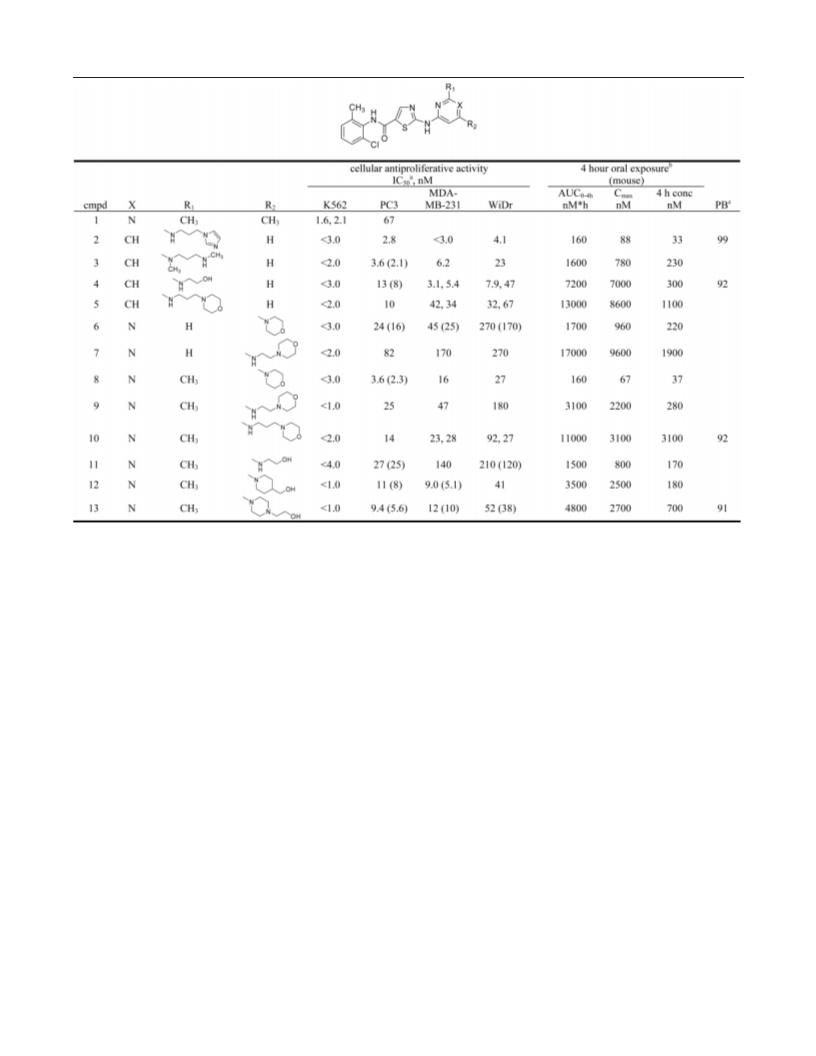
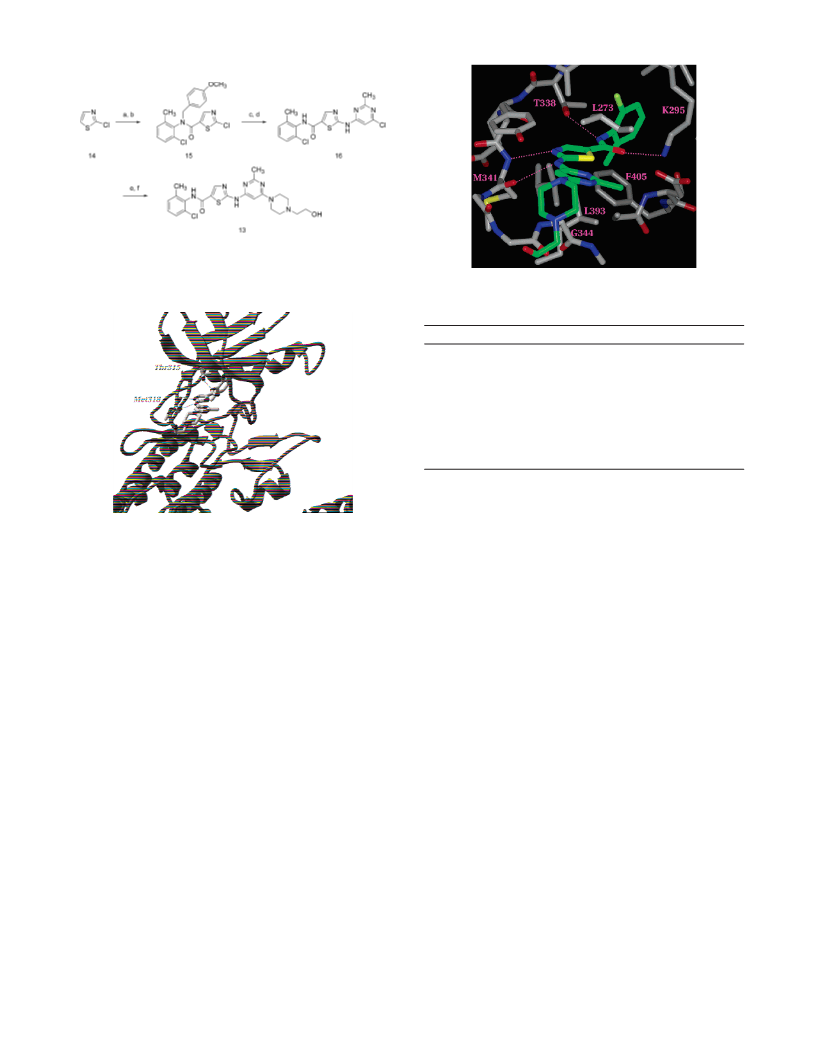
A series of substituted 2-(aminopyridyl)- and 2-(aminopyrimidinyl)thiazole- 5-carboxamides was identified as potent Src/Abl kinase inhibitors with excellent antiproliferative activity against hematological and solid tumor cell lines. Compound 13 was orally active in a K562 xenograft model of chronic myelogenous leukemia (CML), demonstrating complete tumor regressions and low toxicity at multiple dose levels. On the basis of its robust in vivo activity and favorable pharmacokinetic profile, 13 was selected for additional characterization for oncology indications.
View More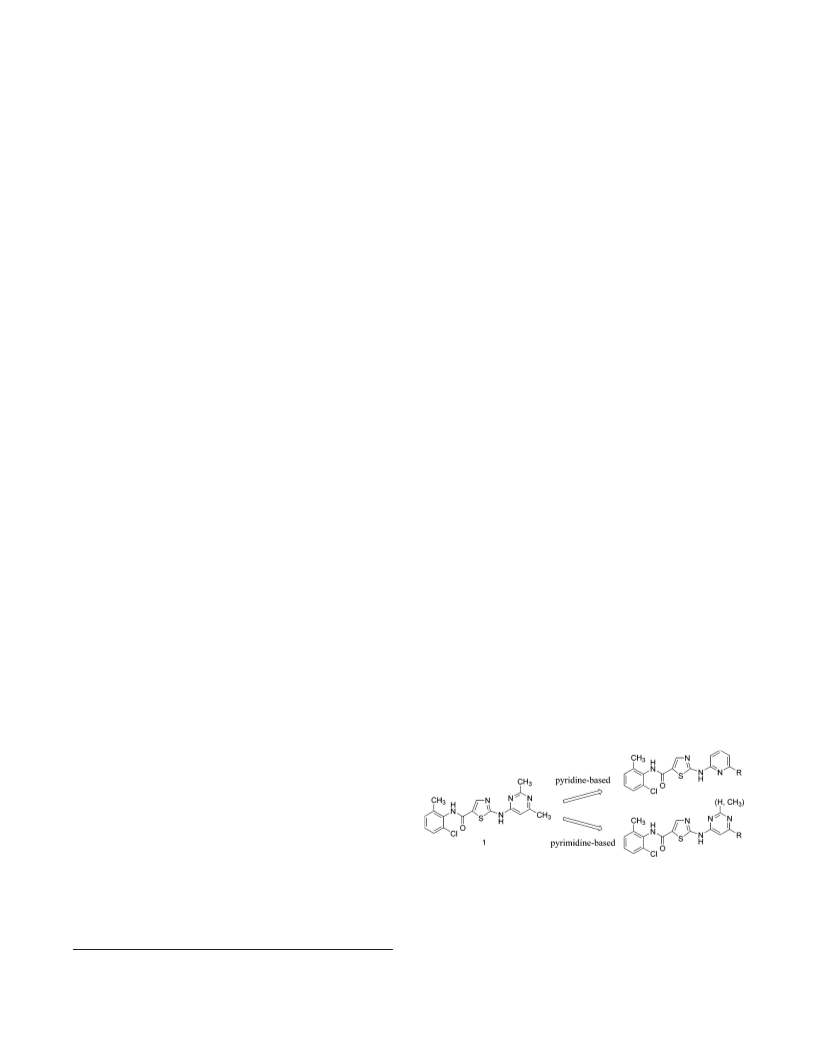

Improve Medical Technology(Nanxiong) Co., Ltd
Contact:86-751-3836997
Address:No.33, Pingan First Road, Fine Chemical Industry Base, Nanxiong City, Shaoguan, Guangdong, China
Liao Cheng All Win Chemicals Co.,LTD
Contact:86+0635-2991582
Address:Room 402,Unit 1,No.27 building.Zhong tong shi dai haoyuan,liaocheng city,Shan dong Province.China
Tianjin Te-An Chemtech Co., Ltd.
Contact:+86-22-65378638
Address:A5-8, No.80 Haiyun Street, TEDA
Contact:+86-838-5655598
Address:Guanghan Nanfeng Industrial Zone
Yuan Shi(SuQian)Biotechnology Co.,Ltd
website:http://www.yuanshibio.com
Contact:+86-527-84226672
Address:jiangsu suqian
Doi:10.1271/bbb.64.1988
(2000)Doi:10.1016/S0040-4020(98)00103-3
(1998)Doi:10.1246/cl.1982.219
(1982)Doi:10.1021/jo300879r
(2012)Doi:10.1021/jo980189l
(1998)Doi:10.1016/j.electacta.2021.138528
(2021)
