Scheme 1
methylsilane as internal standard. Gas chromatography (GC)
analyses were carried out on a Hewlett-Packard HP 5890 gas
chromatograph with a flame ionization detector (FID). The GC
was equipped with a fused silica capillary column (30 m × 0.25
mm i.d) coated with 0.25 µm DB-5 (J &W Scientific, Folsom, CA)
and nitrogen was used as carrier gas. An ELDS laboratory data
system from Chromatography Data System Inc. (Svartsjo¨, Sweden)
was used for registration and processing of the detector signal.
Mass spectrometric (MS) analyses were performed on a Finnigan
Incos 50 quadrapole instrument equipped with a Varian 3400 gas
chromatograph with an on-column injector and a direct insertion
probe. The MS analyses were performed in electron impact (EI)
and positive ion chemical ionization (PCI) modes. Introduction
of the sample into the ion source was made via GC using an on-
column technique. The GC was equipped with a fused silica
capillary column (25 m × 0.25 mm i.d.) coated with 0.2 µm CP-sil
8CB (Chrompack, Middelburg, The Netherlands) and helium was
used as carrier gas. The temperature programming of the GC oven
was as follows: 35 °C for 1.0 min followed by a temperature
increase of 10 °C/min up to 295 °C. The GC-MS transferline was
held at 310 °C. Introduction via the direct insertion probe was
performed at 30 °C followed by a linear temperature increase to
350 °C. The ion source was held at a temperature of 150 °C and
the electron energy was 70 eV in the EI mode. In PCI mode the
ion source was held at 80 °C, the electron energy was 110 eV, and
the ion source pressure was about 1 Torr. At chemical ionization,
NMR (CDCl3): δ 201.03 (CHO), 76.53 (CH2CHO), 71.68, 71.29,
70.17 ((CH2O)3), 31.55 (CH3CH2CH2), 29.56 (5 C:s), 29.45, 29.31
((CH2)7), 26.06 (CH2CH2O), 22.64 (CH3CH2) 14.27 (CH3). MS-DIP-
PCI m/z (% rel int): 273 (M + 1)+ (12), 272 M+ (2), 271 (M - 1)+
+
(2), 243 (M + 1 - 30)+ (4), 229 (M + 1 - 44)+ (7), 169 (C12H25
)
(3), 105 (HO(CH2CH2O)2H)+ (62), 87 (CH2CH2OCH2CHOH)+ (100).
C12H25(OCH2CH2)2OCH2CHO, 3. Yield 28%. FT-IR (neat):
2854 and 2925 cm-1 (C-H), 2715 cm-1 (C-H, aldehyde), 1736
cm-1 (CdO), 1466 cm-1 (C-H in -CH2-), 1122 cm-1 (C-O). 1H
NMR (CDCl3): δ 9.72 (s, 1H, CHO), 4.17 (s, 2H, CH2CHO), 3.74-
3.58 (m, 8H, (CH2O)4), 3.43 (tr, 2H, CH2CH2CH2O), 1.58 (m, 2H,
CH2CH2CH2O), 1.24 (m, 18H, (CH2)9), 0.80 (tr, 3H, CH3). 13C
NMR (CDCl3): δ 203.0 (CHO) 77.08 (CH2CHO), 71.98, 71.64, 71.12,
71.02, 70.40 ((CH2O)5), 31.50 (CH3CH2CH2), 30.03 (5 C:s), 29.88,
29.74 ((CH2)7), 26.00 (CH2CH2O), 23.07 (CH3CH2), 14.51 (CH3).
MS-DIP-PCI m/z (% rel int): 317 (M + 1)+ (25), 316 M+ (14), 315
(M - 1)+ (23), 287 (M + 1 - 30)+ (55), 273 (M + 1 - 44)+ (100),
+
166 (C12H22
)
(51), 131 ((CH2CH2O)3H)+ (30), 87 (CH2CH2OCH2-
CHOH)+ (17).
12H25(OCH2CH2)3OCH2CHO, 4. Yield: 40%. FT-IR (neat):
C
2854 and 2924 cm-1 (C-H), 2720 cm-1 (C-H, aldehyde), 1736
cm-1 (CdO), 1466 cm-1 (C-H in -CH2-), 1122 cm-1 (C-O). 1H
NMR (CDCl3): δ 9.70 (s, 1H, CHO), 4.20 (s, 2H, CH2CHO), 3.75-
3.59 (m, 12H, (CH2O)6), 3.41 (tr, 2H, CH2CH2CH2O), 1.56 (m, 2H,
CH2CH2O), 1.23 (m, 18H, (CH2)9), 0.81 (tr, 3H, CH3). 13C NMR
(CDCl3): δ 201.42 (CHO), 77.26 (CH2CHO), 71.98, 71.61, 71.14,
71.06, 71.02, 70.94, 70.43 ((CH2O)7), 31.52 (CH3CH2CH2), 30.05
(5 C:s), 29.91, 29.77 ((CH2)7), 26.49 (CH2CH2O), 23.11 (CH3CH2),
14.56 (CH3). MS-DIP-PCI m/z (% rel int): 361 (M + 1)+ (7), 360
methane of >99.995% purity was utilized as reagent gas and the
+
instrument was tuned by optimizing the reactant ions (CH5
,
C2H7+, and C3H8+) to an approximate ratio of 5:4:1. The MS scan
range in all analyses was m/z 50-600 and the scan cycle time was
0.6 s (GC introduction) and 1.6 s (DIP introduction).
M+ (2), 359 (M - 1)+ (1), 331 (M + 1 - 30)+ (2), 317 (M + 1 - 44)+
+
(22), 175 ((CH2CH2O)3H)+ (49), 166 (C12H22
)
(28), 131 ((CH2-
CH2O)2CH2CHO)+ (17), 87 (CH2CH2OCH2CHO)+ (100).
Syn th esissDodecylethoxylated Aldehydes (Scheme 1)sA mix-
ture of the appropriate dodecylethoxylated alcohol (C12E1-5OH)
(1 mmol), DMSO (1.2 g, 15 mmol), and DCC (0.62 g, 3.0 mmol) in
toluene (20 mL) was stirred at room temperature for 30 min.
Pyridine (0.080 g, 1.0 mmol) and trifluoroacetic acid (0.060 g, 0.50
mmol) were added to the mixture to generate pyridinium tri-
fluoroacetate.12 The mixture was then stirred for 48 h at room
temperature. Water (10 mL) was added to the reaction mixture,
which was then filtered. Diethyl ether (100 mL) was added and
the organic phase was washed with HCl (3%), saturated aqueous
NaHCO3, and water, dried over MgSO4, and concentrated in a
vacuum. The crude product was chromatographed on a silica gel
column eluted with an increasing content of ethyl acetate 30-
70% in dichloromethane to give the pure aldehydes 1-5 (Scheme
1) as clear oils in 23-40% yield. Identification was performed with
FT-IR, NMR, and MS.
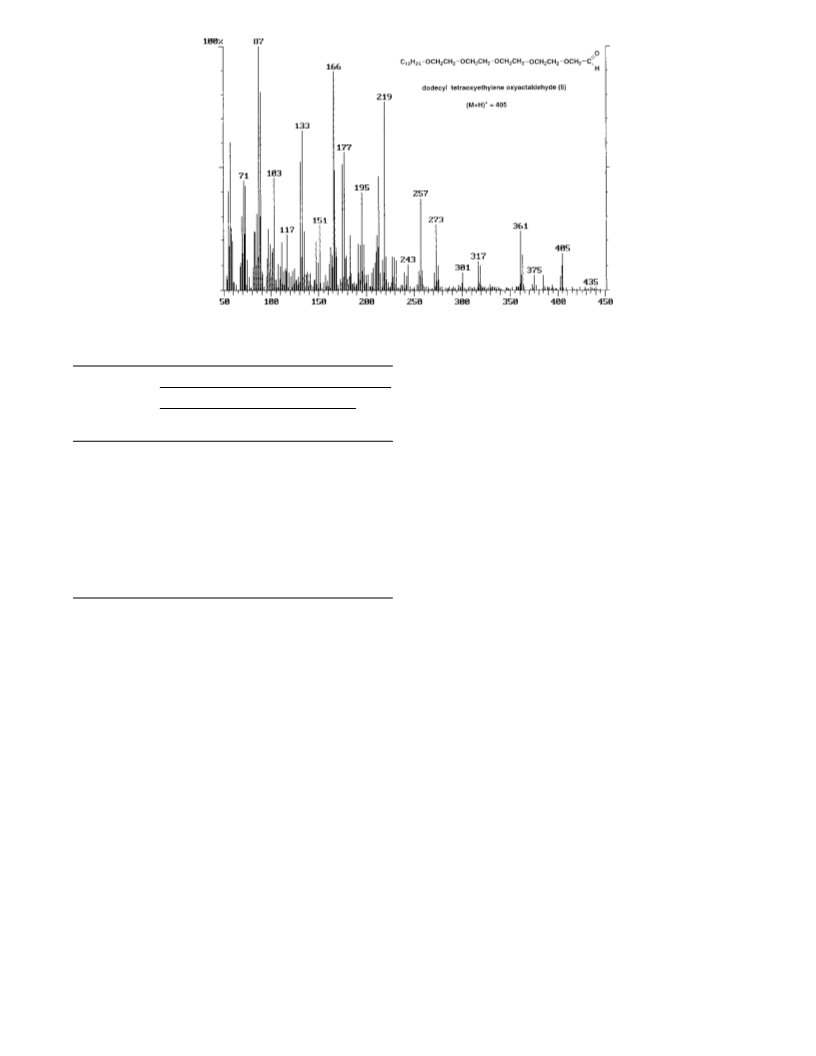
C12H25(OCH2CH2)4OCH2CHO, 5. Yield: 40%. FT-IR (neat):
2834 and 2924 cm-1 (C-H), 2710 cm-1 (C-H, aldehyde), 1736
cm-1 (CdO), 1466 cm-1 (C-H in -CH2-), 1116 cm-1 (C-O). 1H
NMR (CDCl3): δ 9.69 (s, 1H, CHO), 4.13 (s, 2H, CH2CHO), 3.85-
3.52 (m, 16H, (CH2O)8), 3.40 (tr, 2H, CH2CH2CH2O), 1.55 (m, 2H,
CH2CH2O), 1.22 (m, 18H, (CH2)9), 0.84 (tr, 3H, CH3). 13C NMR
(CDCl3): δ 201.13 (CHO), 77.00 (CH2CHO), 71.68, 71.40, 71.19,
71.08, 71.00, 70.89, 70.72, 70.68, 65.98 ((CH2O)9), 31.54 (CH3-
CH2CH2), 29.58 (5 C:s), 29.47, 29.35 ((CH2)7), 26.10 (CH2CH2O),
22.83 (CH3CH2), 14.27 (CH3). MS-DIP-PCI m/z (% rel int): 405
(M + 1)+ (10), 404 M+ (2), 403 (M - 1)+ (4), 375 (M + 1 - 30)+ (4),
361 (M + 1 - 44)+ (28), 219 ((CH2CH2O)4H)+ (73), 175 ((CH2-
CH2O)3H)+ (63), 166 (C12H22
(CH2CH2OCH2CHO)+ (100).
)
+ (91), 133 ((CH2CH2O)2H)+ (50), 87
Dodecyltetraoxyethyleneoxyacetaldehyde and Its Diethyl Acetal
(an Alternative Method for the Synthesis of 5; Scheme 2)sSodium
hydride (60% in mineral oil, 0.22 g, 5.5 mmol) was stirred in DMSO
(dry, 2.0 g) at room temperature for 2 h under nitrogen. Com-
pound 9 (2.0 g 5.5 mmol) was added slowly and the mixture was
stirred at room temperature for 30 min. Bromoacetaldehyde
diethyl acetal (1.1 g, 5.5 mmol) was added dropwise and the
suspension was stirred at room temperature for 48 h. Saturated
aqueous NaHCO3 was added and the mixture was extracted with
ethyl acetate. The organic phase was dried over MgSO4 and
concentrated in a vacuum. The crude product was purified with
flash chromatography on a silica gel column eluted with an
increasing content of ethyl acetate 0-50% in dichloromethane.
Product 6 was obtained in 30% yield.
C
12H25OCH2CHO, 1. Yield: 23%. FT-IR (neat): 2854 cm-1 (C-
H, aliphatic), 2710 cm-1 (C-H, aldehyde), 1740 cm-1 (CdO), 1466
cm-1 (C-H in -CH2-), 1122 cm-1 (C-O). 1H NMR (CDCl3): δ
9.73 (s, 1H, CHO), 4.05 (s, 2H, CH2CHO), 3.52 (tr, 2H, CH2OCH2-
CHO), 1.54 (m, 2H, CH2CH2O), 1.23 (m, 18H, (CH2)9), 0.83 (tr,
3H, CH3). 13C NMR (CDCl3): δ 201.35 (CHO), 76.42 (CH2CHO),
72.38 (CH2OCH2CHO), 31.88 (CH3CH2CH2), 29.76 (6 C:s), 29.71
((CH2) 7), 26.10 (CH2CH2O), 22.81 (CH3CH2), 14.27 (CH3). MS-
DIP-PCI m/z (% rel int): 228 M+ (14), 229 (M + 1)+ (6), 227 (M -
1)+ (38), 199 (M + 1 - 30)+ (100), 185 (C12H25OH)+ (4), 169
+
(C12H25
)
(41), 43 (CH2CHO)+ (38).
C
12H25OCH2CH2OCH2CHO, 2. Yield: 26%. FT-IR (neat): 2854
and 2924 cm-1 (C-H), 2715 cm-1 (C-H, aldehyde), 1736 cm-1
(CdO), 1466 cm-1 (C-H in -CH2-), 1122 cm-1 (C-O). 1H NMR
(CDCl3): δ 9.71 (s, 1H, CHO), 4.13 (s, 2H, CH2CHO), 3.71-3.51
(m, 4H, (CH2O)2), 3.42 (tr, 2H, CH2CH2CH2O), 1.55 (m, 2H,
CH2CH2CH2O), 1.22 (m, 18H, (CH2)9), 0.83 (tr, 3H, CH3). 13C
C
12H25(OCH2CH2)4OCH2CH(OCH2CH3)2, 6. FT-IR (neat): 2948
and 2840 cm-1 (C-H), 1466 cm-1 (C-H in -CH2-), 1150 cm-1
(C-O). 1H NMR (CDCl3): δ 4.57 (tr, 1H, CH(OCH2CH3)2), 3.63-
3.49 (m, 22H, (CH2O)11), 3.40 (tr, 2H, CH2CH2CH2O), 1.52 (m, 2H,
Journal of Pharmaceutical Sciences / 277
Vol. 87, No. 3, March 1998

 Bergh, Margareta
Bergh, Margareta