158
Z. Li et al. / Bioorganic Chemistry 37 (2009) 149–161
relevant deiminase activity directed at the CRGKA cytoplasmic tails
ADs in hope that the S-nitroso-L-homocysteine derived Cys-adduct
of G. lamblia VSPs, cannot be dismissed.
formed using one of these might survive ionization in the mass
spectrometer. The inactivation data observed for BcAD (Fig. 8C
and D) are similar to those determined for GlAD (Fig. 8a and B)
and they define the inactivation constants KI = 66 30 mM and
kinact = 1.0 0.3 minꢀ1. The ESI-MS spectrum of the inactivated
3.8. GlAD substrate specificity
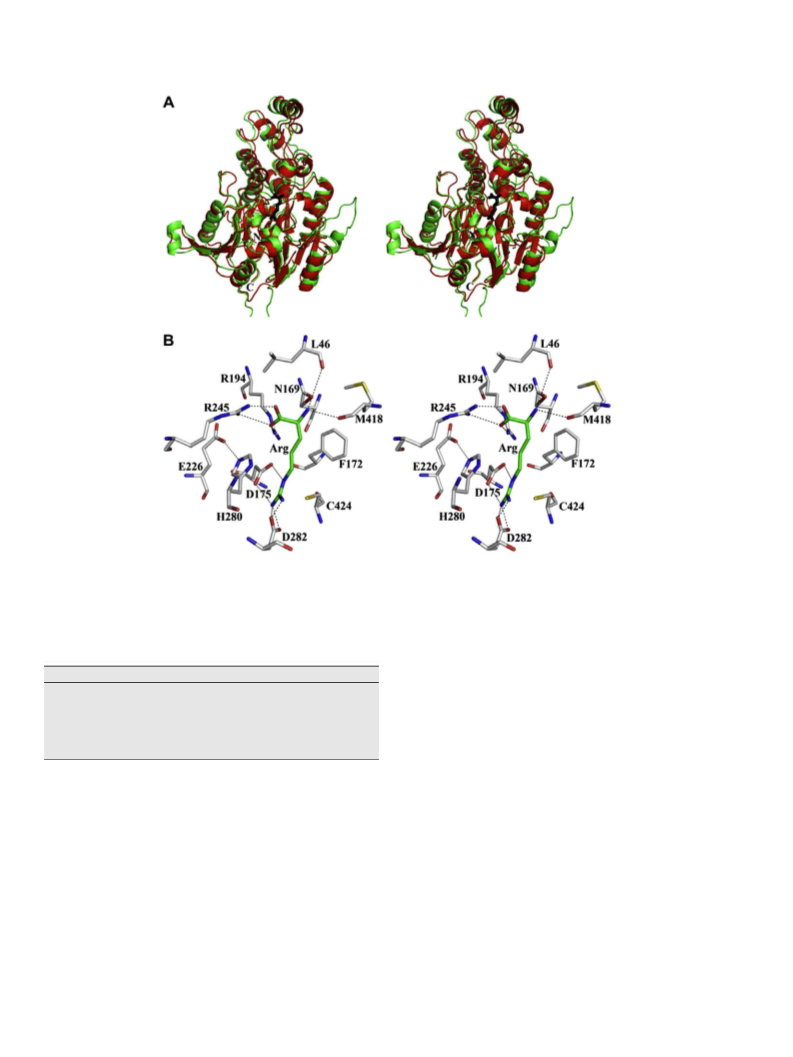
The residues that form the
in the bacterial ADs are conserved in the GlAD active site (see Figs. 2
and 5A and B). In order to characterize GlAD substrate specificity,
L
-Arg CY(COOꢀ)ðNHþ3 Þ binding pocket
(24 lM enzyme incubated 30 min with 10 mM S-nitroso-L-homo-
cysteine in 50 mM K+HEPES pH 7.0, 25 °C) BcAD (native mass
46,638 Da) showed peaks at 46,968 Da (+30 Da), 46,998 Da
(+60 Da), 47,027 Da (+88 Da), 47,131 Da (+193 Da) and 47,162 Da
(+224 Da). Apparently, all three BcAD cysteine residues (each sol-
vent accessible) [30] are subject to modification to form the corre-
sponding S-nitroso adduct (+30 Da) or the N-thiosulfoximide
adduct (+165 Da) (pictured in Fig. 7C).
activity tests were carried out with L-Arg analogs modified at the
CY(COOꢀ)ðNHþ3 Þ moiety. Firstly, agmatine (Arg without the
a-
COOꢀ), shown in a previous study to have no detectable substrate
activity with PaAD [6], was shown in this work to be unreactive with
GlAD. Inhibition studies defined agmatine as a very weak, mixed-
type inhibitor of GlAD: Kis = 50 10 mM and Kii = 120 20 mM.
Arginine ethyl ester on the other hand, undergoes GlAD catalyzed
deimination (kcat = 2.3 sꢀ1 vs. 2.6 sꢀ1for
-Arg) but with a signifi-
cantly increased Km value (2.1 mM vs. 0.16 mM for -Arg). This ana-
L-
In contrast to GlAD and BcAD, S-nitroso-L-homocysteine inacti-
L
vation of DDAH is reported to follow second order kinetics
(k = 4 Mꢀ1 sꢀ1 [35]; k = 9 Mꢀ1 sꢀ1 [37]). X-ray crystallographic
analysis of inactivated DDAH showed that the active site Cys
undergoes N-thiosulfoximide adduct formation and that the sur-
face Cys residue undergoes S-nitrosylation [38].
L
log was also tested as a substrate for EcAD and BcAD and no activity
a
was detected. N -benzoyl-arginine too proved to be an active sub-
strate for GlAD (kcat = 2.5 sꢀ1, Km = 2.3 mM) but not for EcAD or
BcAD. These results suggest that GlAD tolerates mono-alkylation
(but not di-alkylation as in BAEE) at the CY(COOꢀ)ðNH3þÞ units,
whereas its bacterial counterparts do not. Thus, despite the per-
ceived similarity of AD substrate-binding sites, something is clearly
unique about the GlAD site, too subtle to glean from the model, but
nevertheless significant enough to cause a small change in the sub-
strate specificity.
Even though GlAD (and BcAD) binds S-nitroso-
at the active site, the binding is weak compared to substrate-bind-
ing. This is because, without the guanidinium group of the -Arg,
L-homocysteine
L
the favorable ligand binding energy derived from hydrogen bond
interaction with Asp175 and Asp282 and ion pair formation with
the Cys424 thiolate anion is lost (see Fig. 5B). In order to saturate
the active site a high concentration of S-nitroso-L-homocysteine
L
-Homoarginine (
L
-Arg with one additional CH2 unit) is not a
is required and this, in turn, is expected to enhance the rate of col-
lision-based reactions of surface Cys residues. Furthermore, at pH
7.0, the Cys-thiol is protonated. Bound L-Arg dramatically reduces
the pKa of the AD active site Cys-thiol so that it is ionized for nucle-
ophilic addition [17]. Because the S-nitroso group of the S-nitroso-
GlAD substrate (nor a substrate for the bacterial ADs [33,34]) but
rather a weak binding competitive inhibitor (Kis = 48 6 mM). To-
gether, these substrate screening results provide a guide to the de-
sign of mechanism-based inhibitors (see below), and suggest the
requirement that these inhibitors possess
a
L
-CH2CH2CY2CH
L-homocysteine ligand is not positively charged, we anticipate that
(COOꢀ)ðNHþ3 Þ unit to insure effective active site targeting.
its binding will not cause a large reduction in the pKa of the active
site Cys-thiol. Consequently, the protonation state of the active site
Cys-thiol might not differ dramatically from those of the surface
Cys-thiols. Because BcAD possesses two surface Cys residues [30],
it is therefore not surprising that all three BcAD Cys residues are
3.9. Investigation of AD inhibition by active site directed L-Arg analogs
Two designs of Cys-directed GlAD covalent inhibitors are illus-
trated in Fig. 7A and B. One involves the use of an Arg analog to de-
liver an electrophilic ‘‘warhead” to the active site Cys424
nucleophile (Fig. 7A). The other employs an Arg analog, modified
at the guanidinium moiety, that will undergo the first partial reac-
tion but not the second (see Fig. 7B). Both inhibitor designs are
based on earlier studies of mechanism-based inhibitors of the
Cys protease papain.
were modified by the S-nitroso-L-homocysteine.
Attention was next directed to the
(Z)-2-cyanoguanidino-butanoic acid (N -cyano-L-Arg; Fig. 7D). The
L
x
-Arg analog (S)-2-amino-4-
results from previous work had shown that peptide analogs func-
tionalized with a cyano group inhibit papain via formation of the
corresponding thioimidate adducts [39–42]. Thus, one possible
x
pathway for N -cyano-
L-Arg induced GlAD inactivation is nucleo-
The first inhibitor explored is S-nitroso-
L
-homocysteine
philic attack of the GlAD Cys-residue on the cyano carbon to form
the conjugated covalent adduct depicted in Fig. 7D. Alternatively,
nucleophilic addition of the GlAD Cys-thiolate anion at the guanid-
(Fig. 7C). This compound has previously been shown to modify
protein Cys residues, most notably those of mammalian DDAHs,
by nitrosylation and/or by formation of the corresponding N-thio-
inium carbon, followed by loss of ammonium ion might lead to a
x
sulfoximide adduct [35,36]. Accordingly, S-nitroso-
L
-homocysteine
dead-end Cys-thioureido adduct (Fig. 7D). When N -cyano-
L-Arg
(2–90 mM) and GlAD (10
l
M) were incubated in 50 mM K+HEPES
(40 mM) was tested as a GlAD inhibitor neither reversible nor irre-
(pH 7.5, 25 °C). Aliquots were removed, after various incubation
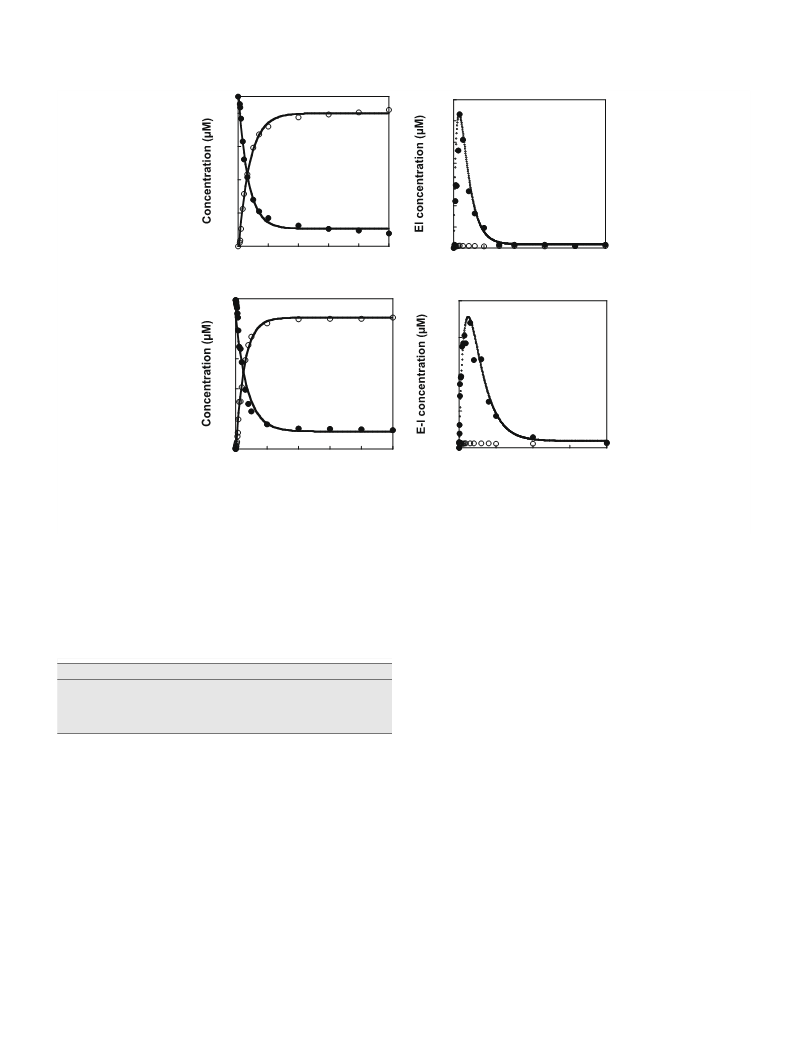
periods, for GlAD activity determination. The value of kobs for inac-
tivation was determined from the slope of the plot of the percent
remaining GlAD activity vs. incubation time (Fig. 8A). The curva-
versible inhibition was observed. On the other hand, incubation of
x
BcAD (6
l
M) with varying concentrations of N -cyano-
L-Arg (0–
40 mM) resulted in time- and concentration-dependent loss of en-
zyme activity (Fig. 8E and F). The pseudo-first-order rate constants
of inactivation (kobs), determined for each inhibitor concentration,
are plotted in Fig. 8F to show that the value of kobs increases with
increasing inhibitor concentrations until BcAD becomes saturated.
The data were fitted to obtain the kinact and KI values of
0.15 0.04 minꢀ1 and 20 10 mM, respectively. The inactivation
was found to be irreversible by employing overnight dialysis
against 50 mM K+HEPES/1 mM DTT at pH 7.5 and 4 °C. However,
analysis of the inactivated enzyme by ESI-MS failed to detect the
ture observed in the plot of kobs vs. S-nitroso-
centration (Fig. 8B) indicates a two-step inactivation sequence in
which the binding of S-nitroso- -homocysteine to GlAD is followed
L-homocysteine con-
L
by covalent modification. The inactivation constants KI = 40
10 mM and kinact = 0.17 0.02 minꢀ1 were determined by data fit-
ting to Eq. (9). The activity of GlAD was not regained following dial-
ysis for 10 h. This finding indicates that irreversible inhibition has
taken place. On the other hand, the covalent adduct formed by
incubation of 10
l
M GlAD with 10 mM S-nitroso-
L
-homocysteine
modified enzyme and therefore, the structural basis for inactiva-
in 50 mM K+HEPES (pH 7.0, 25 °C) for 1 h was not detected by
tion is not known nor is the reason why N -cyano-L-Arg inactivates
x
ESI-MS analysis. We therefore turned our attention to the bacterial
BcAD but not GlAD.

 Li, Zhimin
Li, Zhimin