Z. H. Sun, U. Schwaneberg et al.
one cycle; 958C, 30 s/558C, 30 s/728C, 30 s, 29 cycles; 728C for
3 min, one cycle), Taq DNA polymerase (2.5 U), dNTP mix
(0.20 mm), template (pET42b(+) harboring ADI H404R; 50 ng),
MnCl2 (0.01–0.1 mm), and 5’-TAC ATA TGT CCG CTG AAA AAC AGA
AG-3’ and 5’-GTG CTC GAG TTA GTA GTT GAT CGG-3’ (10 pmol
each) were used. The PCR products were purified by using a QIA-
quick PCR Purification Kit. The purified epPCR products were
cloned into expression plasmid pET42b(+) by MEGAWHOP.[26] For
MEGAWHOP (688C for 5 min, one cycle; 958C for 5 min, one cycle;
958C, 1 min/558C, 1 min/688C, 13 min 30 s, 24 cycles; 688C for
30 min, one cycle), Taq DNA polymerase (1 U), Pfu DNA polymerase
(0.1 U), dNTP mix (0.20 mm) together with template (pET42b(+)
harboring ADI gene H404R; 200 ng) were used. Following the PCR,
DpnI (40 U; New England Biolabs) was added, and the mixture was
incubated (4 h; 378C). The MEGAWHOP products were transformed
into E. coli BL21-Gold (DE3) for expression and screening.
deionized water to ensure the concentration of produced citrulline
was within linear detection range, the solution (400 mL) was mixed
with acid-ferric solution (600 mL) and DAM-TSC solution (100 mL).
The reaction mixture was incubated for a further 30 min at 708C,
followed by incubation in ice water to stop color development.
Absorbance was measured at 530 nm by using a Specord 200
(Analytik Jena AG, Jena, Germany).
Normalization of protein expression for wild-type ADI and variants:
An Agilent Protein 230 Kit (Agilent Technologies Deutschland) and
an Agilent 2100 Bioanalyzer were used to normalize protein
expression in crude cell extract. The protocol used was according
to the Agilent protein 230 Kit Guide, except BSA was used as an
internal standard.
Expression of ADI in a shaking flask and purification: Shaking
flasks (1 L) containing autoinduction medium LS-5052 (200 mL)
supplemented with kanamycin (50 mgmLÀ1) were inoculated with a
1:200 dilution of overnight culture (E. coli BL21-Gold(DE3) harbor-
ing pET42b-ADI) grown in noninducing medium LSG. After 12 h of
expression, E. coli cells were harvested by centrifugation (Eppen-
dorf 5810R 48C, 3220g, 30 min) and resuspended in phosphate
buffer (20 mL, NaxPO4, 20 mm, pH 7.0). E. coli cells were sub-
sequently lysed by using a high-pressure homogenizer (1500 bar,
two cycles; Avestin Emulsiflex, Mannheim, Germany). The disrupted
cells were centrifuged (Eppendorf 5417R, 48C, 13000g, 20 min),
and the supernatant was further cleared by filtration through a
low-protein-binding filter (0.45 mm; Minisart RC 25 single-use sy-
ringe filter; Sartorius, Hamburg, Germany). Wild-type ADI and mu-
tants H404R and K5T/D44E/H404R were subsequently purified by
column chromatography: 1) The filtered cell lysates were subjected
to a Super-Q anion-exchange column that had been pre-equilibrat-
ed with phosphate buffer (NaxPO4, pH 7.0, 50 mm). Cell lysate
(20 mL) was loaded. ADIs were eluted by a NaCl step elution in
Cultivation and expression in 96-well plates: Colonies grown on
LBkan agar plates were transferred, by using toothpicks, into 96-well
microtiter plates (flat-bottomed, polystyrene plates; Greiner Bio-
One GmbH, Frickenhausen, Germany), containing LSG noninducing
medium (150 mL)[27] supplemented with kanamycin (50 mgmLÀ1).
After 16 h of cultivation in a microtiter plate shaker (Multitron II,
Infors GmbH, Einsbach, Germany; 378C, 900 rpm, 70% humidity),
each well was replicated by using a replicator (EnzyScreen BV,
Leiden, Netherlands) into a second series of 96-well microtiter
plates containing autoinduction medium LS-5052 (150 mL)[27] sup-
plemented with kanamycin (50 mgmLÀ1). The first set of plates was
stored at À808C after addition of glycerol. The clones in the
second set of plates were cultivated for 12 h (Multitron II, Infors
GmbH, 378C, 900 rpm) and used for screening.
Screening procedure
phosphate buffer (NaxPO4, pH 7.0, 50 mm) at a rate of 3 mLminÀ1
.
96-well plate-format citrulline colorimetric screening assay: A modi-
fied protocol for citrulline detection based on the carbamido-diace-
tyl reaction[28] (see Scheme 1 for assay mechanism) was used to
measure the activity of ADI. Screening for increased activity was
carried out by measuring the activities at pH 7.4 and 6.4.
2) The ADI protein obtained by ion-exchange chromatography was
subjected to a gel filtration column (Matrix: Toyoperl HW-55S,
buffer: NaxPO4, pH 7.4, 50 mm, bed volume: 33 mL, bed height:
40 cm, column: Omnifit).
Cell culture (20 mL) was transferred into 96-well microtiter plate.
The enzyme reaction was initiated by the addition of arginine solu-
tion (100 mL, 100 mm) supplemented with cetyltrimethylammoni-
um bromide (CTAB, 4 mm), and the mixture was incubated (20 min,
378C). Subsequently, acid-ferric solution (60 mL) and diacetyl mon-
oxime (DAM, 20 mL, 0.5m) were added. The reaction mixture was
incubated for a further 15 min at 558C. Absorbance was measured
at 492 nm on a microtiter plate reader (Tecan Sunrise, Tecan Group
AG, Zꢁrich, Switzerland).
Purified ADI was subsequently concentrated in a Amicon ultra-4
centrifugal filter device (Millipore) with a 30 kDa cut-off membrane.
The total protein concentration was determined by BCATM assay
kit (Pierce, Born, Germany), and the homogeneity of the purified
sample was controlled by SDS-PAGE by using standard molecular
biology techniques.
Characterization of wild-type ADI and mutants
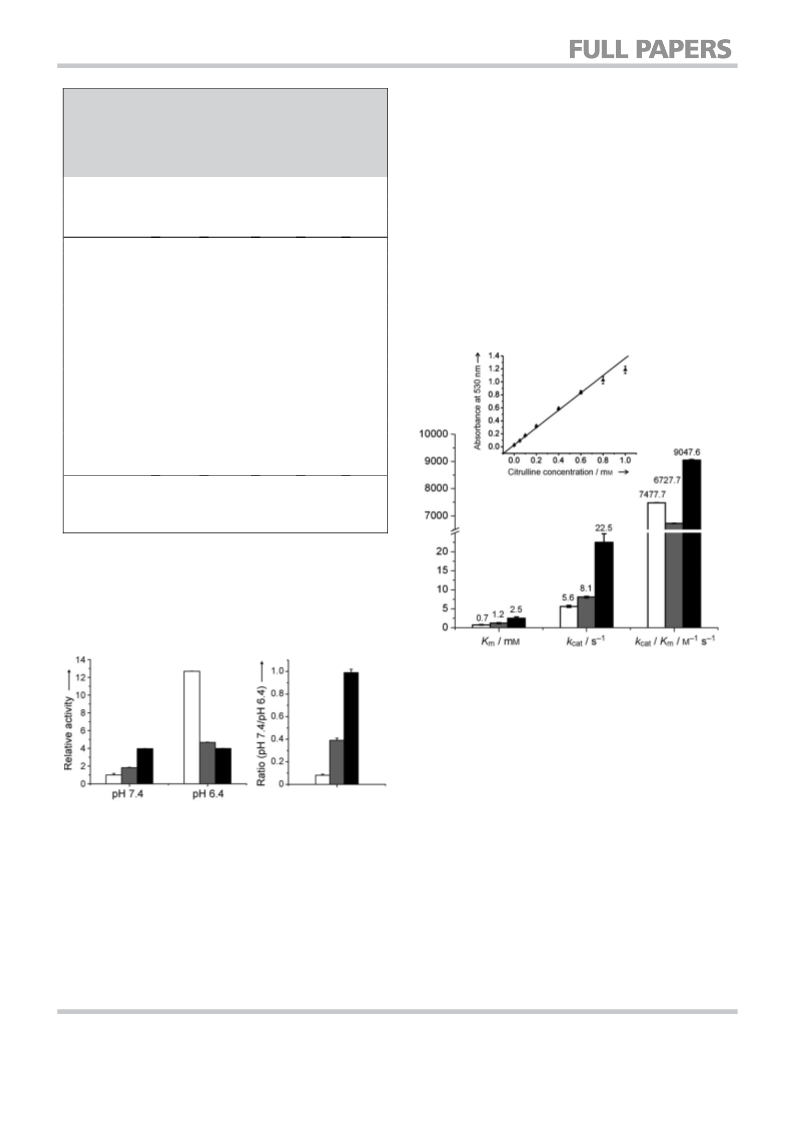
Determination of kcat and Km of wild-type ADI and the mutants: The
kcat and Km values were determined from initial-velocity data mea-
sured as a function of substrate concentration. Enzyme reactions
were carried out at 378C in a water bath. After 10 min of preincu-
bation at 378C, the reaction was initiated by addition of purified
enzyme (50 mL, 0.2–0.5 mm) to the substrate solution (200 mL, 0.2–
10 mm of arginine, 0.5m phosphate buffer, pH 7.4) in deep-well
plates. The reaction mixture was incubated at 378C, and every
2 min an aliquot (30 mL) was transferred from each well to acid-
ferric solution (30 mL) to stop the enzyme reaction. The color devel-
opment was subsequently performed in a 96-well PCR plate.
Ferric-acid solution (90 mL) and DAM-TSC solution (15 mL) were
added to each well. The 96-well PCR plate was then incubated at
708C for 30 min in a thermal cycler (Eppendorf Mastercycler gradi-
ent) for color development, followed by incubation in ice water to
stop color development. Absorbance was measured at 530 nm by
using a microtiter plate reader (SPECTROstar Omega, BMG Labtech,
Standard-deviation measurements were performed in 96-well plate
format by using culture from BL21-Gold (DE3) lacking ADI and in a
separate experiment containing ADI. Apparent standard deviation
was based on the absolute absorbance values obtained from the
ADI wild-type plate. The true standard deviation was calculated by
subtracting the background absorbance value of the microtiter
plate lacking ADI from the apparent values.
Cuvette-format citrulline colorimetric assay: ADI activity was routine-
ly measured by using a modified citrulline-detection protocol with
DAM and TSC[29] (see Figure 1A for assay mechanism) in Eppendorf
tubes. The enzyme reaction was initiated by the addition of argi-
nine solution (200 mL, 100 mm) to a 2 mL Eppendorf tube contain-
ing crude cell lysate (50 mL), and the mixture was incubated for
20 min at 378C. Subsequently, acid-ferric solution (250 mL) was
added to stop the enzyme reaction. After appropriate dilution with
696
ꢀ 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
ChemBioChem 2010, 11, 691 – 697

 Zhu, Leilei
Zhu, Leilei