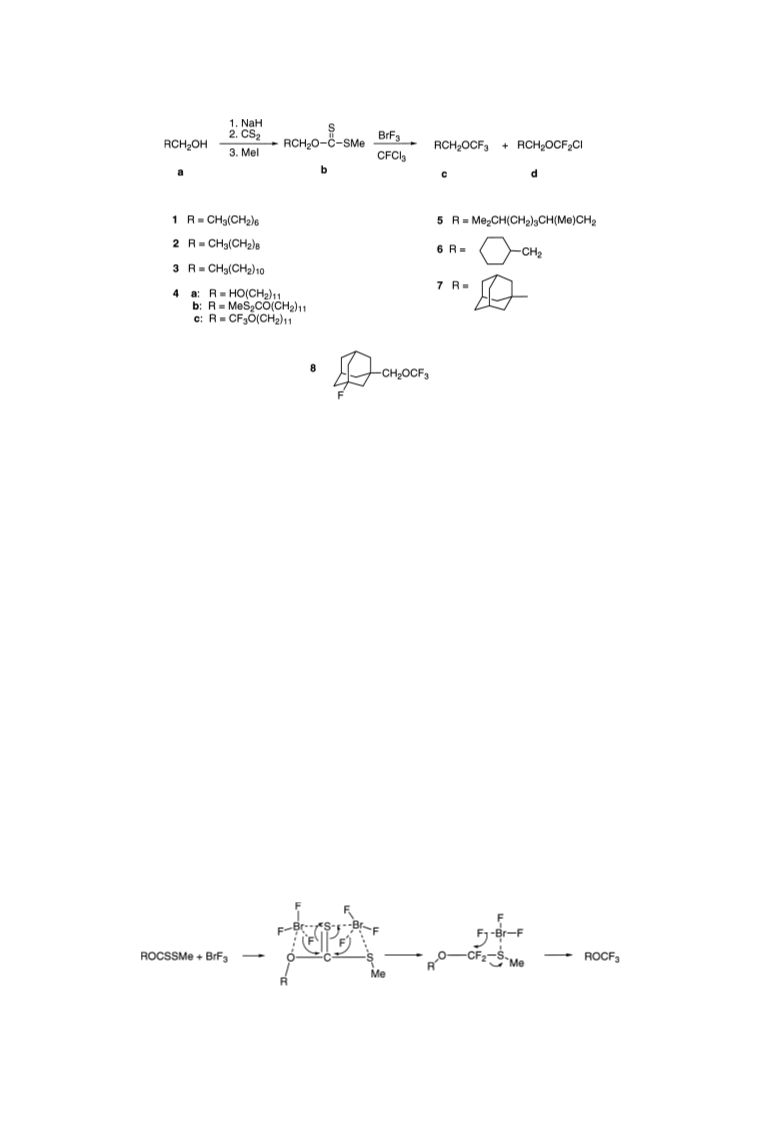
I. Ben-David et al. / Journal of Fluorine Chemistry 97 (1999) 75±78
77
3. Experimental
aqueous Na2SO3 until it was colorless, neutralized with
aqueous NaHCO3, extracted with CH2Cl2, dried over
MgSO4 and evaporated. The products were puri®ed by
fractional distillation.
1H NMR spectra were recorded with Bruker AC-200 and
AM-360 WB spectrometers, with CDCl3 as solvent and
Me4Si as an internal standard. The 19F NMR spectra were
measured at 338.8 MHz and are reported in parts per million
up®eld from CFCl3, which also served as an internal
standard. IR spectra were recorded as neat ®lms, in CHCl3
solution or in KBr pellets on a Nicolet 205 FTIR spectro-
photometer.
Octyl tri¯uoromethyl ether (1c): 15 mmol of 1b in 250 ml
of CFCl3 was reacted with 45 mmol (2.5 ml) of BrF3 in
100 ml of CFCl3 according to the general procedure
described above. Compound 1c was isolated as a colorless
oil, b.p.(171 mm) 95±978C in 82% yield. 1H-NMR: 3.94 (2H,
t, J6.5 Hz), 1.68 (2H, quin., J6.6 Hz), 1.5±1.2 (10H, m),
0.89 (3H, t, J6.5 Hz). 19F-NMR: 61.13 (s). 13C-NMR:
122 (q, J251 Hz), 67, 32, 29, 28, 25, 23, 14. MS (m/e): 198
3.1. Preparation and handling of BrF3
[(M) ], 99 (CH2OCF3) , 69 (CF3). Anal. Calcd. for
C9H17F3O: C, 54.53%; H, 8.64%. Found: C, 54.58%; H,
8.62%.
Although commercially available, we prepare our own
BrF3 by simply passing 0.58 mol of pure ¯uorine through
0.2 mol of bromine placed in a copper reactor at 0±108C.
When no excess of bromine is present the BrF3 obtained is
an inde®nitely stable pale yellow liquid and has a density of
2.5 with a m.p. of 7±98C [19]. At this temperature, the
higher oxidation state derivative (BrF5), will not form in any
appreciable amount [20], although we always use a small
excess of bromine thereby keeping the reagent from dis-
proportionation to BrF5. This is also responsible for the
reddish coloration of the reagent. We store the reagent in
te¯on containers for long periods. BrF3 is a strong oxidizer
and tends to react very exothermically with water and
oxygenated organic solvents. The work with BrF3 should
be conducted in a well-ventilated area and caution and
commonsense should be exercised.
Decyl tri¯uoromethyl ether (2c): 10 mmol of 2b in 250 ml
CFCl3 was reacted with 30 mmol (1.67 ml) of BrF3 in
100 ml CFCl3 according to the general procedure described
above. Compound 2c was isolated as a colorless oil,
1
b.p.(7 mm) 79±828C in 89% yield. H-NMR: 3.93 (2H, t,
J6.4 Hz), 1.67 (2H, br quin., J6.6 Hz), 1.5±1.0 (14H, m),
0.87 (3H, t, J6.3 Hz). 19F-NMR: 61.29 (s). Anal. Calcd.
for C11H21F3O: C, 58.39%; H, 9.35%; F, 25.19%. Found: C,
58.27%; H, 9.26%; F, 24.78%.
Dodecyl tri¯uoromethyl ether (3c): 2 mmol of 3b in 25 ml
of CFCl3 was reacted with 6 mmol (0.33 ml) of BrF3 in
15 ml CFCl3 according to the general procedure described
1
above. 3c (oil) was obtained in 80% yield. H-NMR: 3.94
(2H, t, J6.6 Hz,), 1.4±1.1 (20H, m), 0.88 (3H, t,
J6.5 Hz). 19F-NMR: 61.20 (s). High resolution MS
3.2. General procedure for the preparation of aliphatic
xanthates [8]
(m/e): Calcd. for C13H25F3O: 254.1857 [(M )]. Found:
254.1866. Anal. Calcd. for C13H25F3O: C, 61.39%; H,
9.91%. Found: C, 61.12%; H, 9.72%.
A solution of alcohol (30 mmol), sodium hydride
(40 mmol) and imidazole (60 mg) in dry THF (120 ml)
was re¯uxed for 3 h under nitrogen. After cooling to room
temperature, 9 ml of CS2 was added dropwise followed by
30 min re¯ux. The reaction mixture was cooled again to
room temperature, 9 ml of MeI was added dropwise and
re¯uxed for an additional 30 min. The large excess of CS2
and MeI help to speed the reaction. It was then neutralized
by addition of acetic acid, washed with water, extracted with
chloroform and the combined organic layers dried and
evaporated. The product was obtained in 90±97% yield,
the main by-product being dimethyl trisulfur carbonate
(yellow), which does not interfere with the subsequent
reaction.
1,12-Dodecanediol bis(tri¯uoromethyl) ether (4c):
9 mmol of 4b in 250 ml CFCl3 was reacted with 54 mmol
(3 ml) of BrF3 in 100 ml CFCl3 according to the general
procedure described above. Compound 4c was isolated as a
colorless oil, b.p.(17 mm) 95±1008C in 78% yield. 1H-NMR:
3.93 (4H, t, J6.6 Hz), 1.68 (4H, m), 1.5±1.1 (16H, m).
19F-NMR: 61.29 (s). Anal. Calcd. for C14H24F6O2: C,
49.70%; H, 7.15%. Found: C, 50.10%; H, 7.14%.
3,7 Dimethyloctyl tri¯uoromethyl ether (5c): 12 mmol of
5b in 250 ml CFCl3 was reacted with 36 mmol (2 ml) of
BrF3 in 100 ml CFCl3 as described above. Compound 5c
was isolated as a colorless oil, b.p.(17 mm) 758C in 90% yield.
1H-NMR: 3.99 (dt, J17.0 Hz, J21.06 Hz, 2H), 1.8±1.4
(m, 4H), 1.4±1.1 (6H, m), 0.90 (d, J6.9 Hz, 3H), 0.87 (d,
J6.8 Hz, 6H). 19F-NMR: 61.29 (s). 13C-NMR: 121.67
(q, J252), 65.88, 39.12, 36.98, 35.58, 29.24, 27.91, 24.52,
3.3. General procedure for the reaction of xanthates with
BrF3
22.59, 19.22. MS (m/e): 99 ꢁCH2OCF3 , 85 ꢁOCF3 , 69
(CF3). Anal. Calcd. for C11H21F3O: C, 58.39%; H, 9.35%.
Found: C, 58.79%; H, 9.60%.
The xanthates (2±4 mmol) were dissolved in 30±60 ml
dry CFCl3 and cooled to 08C. A 3 mole equivalent of BrF3
was dissolved in 30 ml of the same solvent, cooled to 08C
and added dropwise to the xanthate solution over a period of
5±10 min. The reaction mixture was then washed with
2-Cyclohexylethyl tri¯uoromethyl ether (6c): 6.5 mmol of
6b in 100 ml CFCl3 was reacted with 19.5 mmol (1.1 ml) of
BrF3 in 80 ml CFCl3 as described above. Compound 6c was
isolated as a colorless oil, b.p.(155 mm) 90±928C in 84%

 Ben-David, Iris
Ben-David, Iris