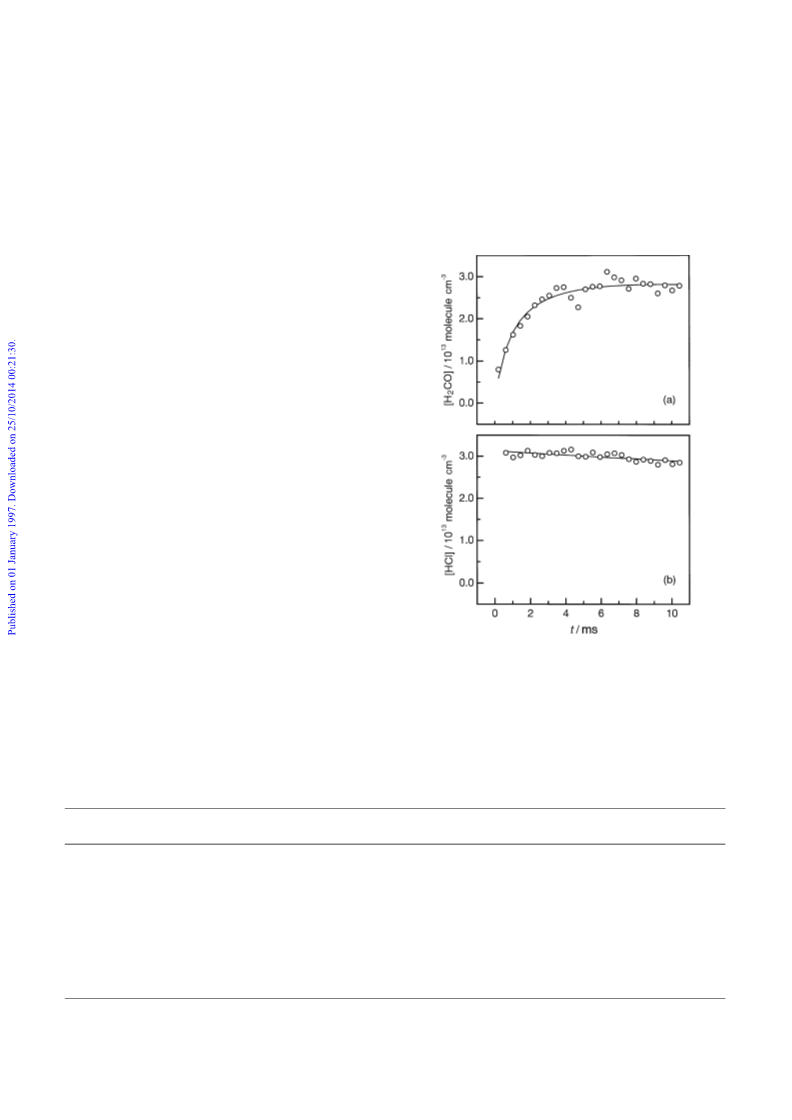
observed data set by more than a factor of two then the depe-
out unequivocally the possibility of the CH SCH O ] O
3
2
2
dence of k on the initial concentration of photolyzed DMS
reaction occurring to some degree. Nevertheless, both their
results and ours provide strong evidence supporting the
notion that, under atmospheric conditions, the fate of the
CH SCH O intermediate is decomposition to CH S and
4
was considered statistically signiÐcant compared to the varia-
bility of k in the observed data set and was thus rejected.
4
Based on the comprehensive battery of numerical simula-
3
2
3
tions described above, we have concluded that secondary
H CO, as opposed to reaction with molecular oxygen.
2
chemistry involving CH S and CH radicals generated via
A complete picture detailing the fate of CH SCH radicals
3
3
3
2
DMS photolysis and CH S radicals produced by the decom-
for the range of tropospheric conditions still requires detailed
knowledge of other potentially important reactions involving
the CH SCH O
3
position of CH SCH O may have made a limited contribu-
3
3
2
tion to the CH SCH O disappearance kinetics observed in
2 2 radical. In particular, reaction with HO2
2 2
3
our experiments. Our results indicate that the true value of the
CH SCH O self-reaction rate coefficient lies within the range
may be important. To our knowledge, there exist no published
measurements of the rate coefficient for the reaction of
CH SCH O with HO . Based on analogy with the reactions
3
2 2
(
1.2 ^ 0.5) ] 10~11 cm3 molecule~1 s~1.
It is worth noting that the secondary chemistry discussed
3
2 2
2
of alkyl peroxyl radicals with HO , one may estimate that the
2
above could not have a†ected the results of our study of the
room-temperature rate coefficient for the CH SCH O
3
2 2
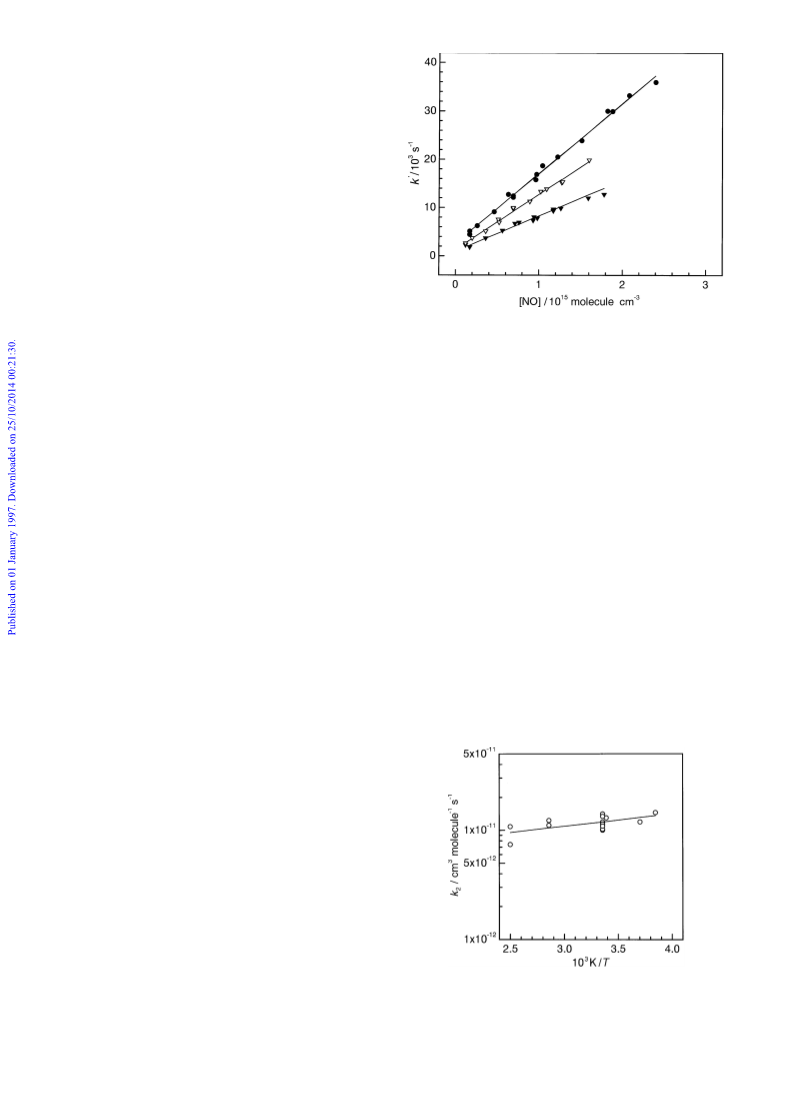
CH SCH O ] NO reaction. This is because peroxyl radical
] HO reaction is of the order of 1 ] 10~11 cm3 molecule~1
3
2 2
2
concentrations were far too low for these species to compete
with NO for consumption of CH SCH O under the experi-
s~1 .22h24 A reasonable remote marine boundary layer diur-
nally averaged HO concentration may be ca. 1 ] 108 mol-
3
2 2
2
mental conditions employed, and because CH S was effi-
ciently scavenged by NO.
ecule cm~3 33 although, of course, actual HO mixing ratios
3
2
in the marine boundary layer will vary widely as a function of
The experiments focusing on the CH SCH O self-reaction
solar Ñux, water vapour, temperature, and the concentrations
of various radical and non-radical species. Using the afore-
3
2 2
have demonstrated that this reaction, like the CH SCH O
2 2
NO reaction, produces H CO, and hence CH S, in approx-
3
]
mentioned HO concentration and rate coefficient one may
2
3
2
imately unit yield, presumably via the decomposition of the
roughly estimate the remote marine boundary layer lifetime of
unstable CH SCH O intermediate. The room-temperature
rate coefficient for the self-reaction is surprisingly fast,
CH SCH O with respect to reaction with HO to be of the
3
2
3
2 2
2
order of 1000 s. Assuming a typical remote marine boundary
layer NO mixing ratio of pptv,15 the lifetime of
CH SCH O with respect to reaction with NO is ca. 700 s.
(
1.2 ^ 0.5) ] 10~11 cm3 molecule~1 s~1. This result is some-
5
what higher than the value of (7.9 ^ 1.4) ] 10~12 cm3
molecule~1 s~1 reported by Wallington et al.13 from their
observation of CH SCH O disappearance. As in our study,
Wallington et al.13 note that the rate coefficient observed in
their study may not be the true CH SCH O self-reaction rate
3
2 2
Therefore, it is entirely possible that in the marine boundary
layer these two reaction pathways are competitive. This is an
important point, as the product of the HO ] CH SCH O
3
2 2
2 3 2 2
reaction is likely a hydroperoxide (CH SCH OOH) that
3
2 2
3
2
coefficient. Wallington et al.13 suggest that complicating sec-
would be susceptible to heterogeneous removal from the
atmosphere via condensation on pre-existing aerosols and
cloud droplets as well as deposition at the ocean surface.
Thus, CH SCH O reaction with HO may be part of an effi-
ondary chemistry, in particular the CH SCH O ] CH S
3
2 2
3
2 2
reaction, may have contributed to the observed CH SCH O
3
decay.
3
2 2
2
cient process that removes volatilized sulfur from the atmo-
sphere, precluding its participation in aerosol and
cloud-formation processes that may impact the earthÏs radi-
ation budget.
Conclusions
We have demonstrated that the reaction of CH SCH O with
2 2
NO, as well as the CH SCH O self-reaction, produce H CO
2 2
and CH S in high yield, and that reaction of the CH SCH O
3
Typically, the self reactions of peroxyl radicals other than
HO and CH O are unimportant in the atmosphere.
3
2
3
3
2
2
3 2
intermediate with O does not compete with its decomposi-
tion under our experimental conditions. However, the cer-
However, given the rather fast rate coefficient for the
CH SCH O self-reaction and the low NO levels typical of
2
3
2 2
tainty of our Ðndings with respect to atmospheric conditions
is a bit more tenuous. We have demonstrated that the lifetime
of the CH SCH O intermediate is less than 30 ls at 261 K
the remote marine boundary layer, it is not out of the ques-
tion that this reaction could be important under certain con-
ditions. An accurate assessment of this reactionÏs potential
atmospheric importance is highly dependent on understand-
ing the kinetics of the CH SCH O ] HO reaction.
3
2
and 10 Torr total pressure. If k , the rate coefficient for
3
CH SCH O decomposition, is assumed to have a value of ca.
5 000 s~1 at atmospheric pressure and room temperature
probably a poor assumption) then in 1 atm of air the
3
2
3
2 2
2
3
The present laboratory study, when combined with other
recent studies,11,13 demonstrates that the atmospheric oxida-
tion of the CH SCH radical will produce the key species
(
CH SCH O ] O rate coefficient need only be ca. 7 ] 10~15
3
2
2
3
2
cm3 molecule~1 s~1 in order to compete equally with
CH SCH O decomposition. Analogous RO ] O reactions
CH S in near unit yield, given the absence of competing reac-
3
tions, such as CH SCH O ] HO . In the light of the pre-
3
2
2
3
2 2
2
have room-temperature rate coefficients of 1.9 ] 10~15 cm3
ceeding discussion, it is apparent that laboratory experiments
designed to assess the kinetics and mechanism of the
CH SCH O ] HO
molecule~1 s~1 for R \ CH
and 1.0 ] 10~14 cm3
3
molecule~1 s~1 for R \ C H .21 It is thus not out of the
2 reaction should be considered a high
2
5
3
3
2 2
question that the reaction CH SCH O ] O could be impor-
priority.
2
2
3
tant in the atmospheric oxidation of CH SCH . However,
2
because the upper limit placed on the CH SCH O lifetime by
This research was supported by the National Science Founda-
tion under grant number ATM-94-12237. Shawn P. Urbanski
was supported by the NASA Earth System Science Fellowship
Program. We would like to thank Tim Wallington for insight-
ful comments and suggestions.
3
2
our experiments is for 10 Torr total pressure and 261 K, it is
highly probable that at atmospheric pressure and tem-
peratures more typical of the lower troposphere, the
CH SCH O lifetime is signiÐcantly less than 30 ls. Addi-
3
2
tionally, while most of the CH S yield experiments of
3
Turnipseed et al.11 were conducted in 22 Torr of O , they
References
2
also conducted some experiments in 200 Torr O and saw no
2
1
H. Berresheim, P. H. Wine and D. D. Davis, in: Composition,
Chemistry and Climate of the Atmosphere, ed. H. B Singh, Van
Nostrand Reinhold, New York, 1995, pp. 251È307, and references
therein.
indications of a CH SCH O ] O reaction producing HO .
3
2
2
2
However, owing to the complexity of their experimental tech-
nique for inferring HO production, they were not able to rule
2
2818
J. Chem. Soc., Faraday T rans., 1997, V ol. 93

 Urbanski, Shawn P.
Urbanski, Shawn P.