8998 J. Agric. Food Chem., Vol. 53, No. 23, 2005
Moraru and Lee
and to compare its stability to the stability of the all-trans form,
when subjected to gastric pH at physiological temperature in a
tributyrin model system. The purified compounds were used to
collect kinetic data, enabling the calculation of reaction rate
constants for the isomerization reactions undergone by the
predominating isomers. Thus, this study attempts to improve
the understanding of pH-driven lycopene isomerization in ViVo,
enabling the prediction of the extent of isomerization based on
gastric residence time.
The HPLC equipment consisted of a 600E solvent delivery system
and 991 photodiode array detector (Waters Corp., Milford, MA) and a
computer running the dedicated Waters 991 PDA software. For faster
processing, the signal at 471 nm, the absorption maxima for lycopene
(13), was exported via an analogue output channel to a second computer
running the Diamir chromatography software (JMBS, Bear, DE).
Preparation of the cis-Lycopene Isomer. To improve the cis
isomers proportion, isomerization of stock lycopene was forced by
placing aliquots of the stock (concentration approximately 120 mg/L)
in a 1.5-mL brown, septum-closed GC vial (National Scientific, Duluth,
GA), and preheating the vial by submersion in an oil bath for 5 min at
8
5 °C (oil temperature). all-trans-Lycopene and the predominating cis-
MATERIALS AND METHODS
lycopene isomer were isolated from the preheated stock by cooling it
down and directly injecting the concentrated stock in the HPLC, to
increase the concentration of the isomers collected (16).
The HPLC system used for the preparation of larger amounts of
lycopene isomers was the system described above, except it used a
Preparation of the Lycopene Stock. The procedure was derived
from procedures reported in the literature (10-12), and was performed
in aluminum foil-wrapped glassware or, when not possible, in dim light.
All reagents were purchased from Fisher Scientific (Fairlawn, NJ), and
solvents were HPLC grade.
2
50 × 10 mm i.d., 5 µm, Develosil RP-Aqueous C30 semi-preparative
Tomatoes for fresh consumption, purchased from a local supermarket
column protected by a semi-preparative scale C18 SecurityGuard
column (both provided by Phenomenex Inc., Torrance, CA), both kept
at 25 °C. The injection size was 100 µL. A fraction collector was
connected to the outlet of the HPLC and programmed to collect every
0.2 min starting at 13 min. To avoid isomerization caused by
temperature or light, fractions were collected in tubes wrapped in
aluminum foil and at the end of each injection were transferred into
larger, septum-closed, amber vials stored in the freezer. Every 4 h, the
similar fractions were pooled together in brown bottles, sealed, and
stored in a low-temperature freezer (-65 °C) until further use. For
longer storage, solvent was removed by flushing with nitrogen in dim
light on ice.
Identification of the Lycopene Isomers Prepared. The fractions
carrying lycopene isomers were tentatively identified spectrophoto-
metrically: 1 mL of each fraction was placed in a quartz cuvette (Fisher
Scientific, Fairlawn, NJ) and its absorption spectrum was read between
300 and 550 nm, at a rate of 100 nm/min, using a U 3110 UV/vis
spectrophotometer (Hitachi Instruments, San Jose, CA). The fractions
containing lycopene isomers were further analyzed with the analytical
HPLC method, which allowed pooling together neighboring fractions
showing the same pattern.
(Shop-Rite, Elizabeth, NJ), were used as the source for preparing all-
trans-lycopene. A 30-g portion of tomatoes was washed, wiped dry,
and then cut in half, and the cores were removed. The material was
mixed with 100 mL of extraction solvent (hexane:acetone:ethanol, 2:1:
1
, v/v/v) in a Nova I blender (Waring Corp., New Hartford, CT). To
prevent heating, mixing was performed for 10 min in an iced water
bath, in 1-min steps alternating with 3-min breaks. After filtration
through Whatman #1 paper (Whatman Inc., Clifton, NJ), the washing
and filtration of the retentate with the same extraction solvent mixture
was repeated until colorless. Filtrates were pooled, and the nonpolar
phase was separated in a separatory funnel. Saponification of the
nonpolar phase with 10% KOH in methanol was performed overnight,
in the dark. The mixture was then rewashed and the nonpolar phase
was again separated in a separatory funnel. After filtration and removal
of solvents using a rotary evaporator at 35 °C water bath temperature,
the preparation was solubilized in 2 mL of hexane:dichloromethane
(4:1, v/v), leading to the crude extract.
Purification was performed using a 300 × 11 mm chromatography
column (Kontes, Vineland, NJ), packed with a 1:1 (w/w) mixture of
magnesium oxide and Celite and a top layer of anhydrous sodium sulfate
of approximately 1-cm height. The sample was loaded onto the column
and then eluted stepwise. The first step used 100-150 mL of petroleum
ether, aiming to remove the more nonpolar species, including lipid
residues. When the coloration of the phase leaving the column returned
from yellow to colorless, petroleum ether with 4% acetone was added.
This caused a slow movement of the intense red band consisting of
lycopene. Elution was stopped when the phase leaving the column was
colorless and the intense red band reached the lower third of the column.
The packing of the column was carefully taken out, and the intensely
red band was separated. The purified lycopene was obtained by
extraction from this area, using the same extraction solvent as above.
After solvent removal, lycopene was redissolved in 2 mL of hexane:
dichloromethane mixture (4:1, v/v), placed in brown GC vials, slightly
flushed with nitrogen, and then sealed and placed immediately in a
low-temperature freezer (-65 °C).
The structural identity of the fractions containing the main two
1
isomers was confirmed using H NMR. For this purpose, the solvent
was removed by flushing with nitrogen, in the dark and on ice, and
then the dried residue was redissolved in deuterated chloroform (17).
The samples were analyzed using a Varian 400
UNITY
INOVA NMR
spectrophotometer with the VNMR version 6.1B (Varian NMR
Systems, Palo Alto, CA) software package, at room temperature. The
spectra obtained were compared with published NMR spectra of
lycopene isomers (18), and the identity of isomers was elucidated by
comparing the value of the spectroscopic shifts.
Kinetic Studies of Lycopene Stability in a Model System, at
Gastric pH. Lycopene is solid and hydrophobic, so a hydrophobic
model system able to dissolve lycopene was necessary. Criteria such
as liquid state, ability to dissolve lycopene, stability at temperatures
above 100 °C, lack of rancidity or oxidation, and relatively low viscosity
were used to select the system. The system of choice was tributyrin, a
liquid triglyceride having the residues of the saturated butyric acid
esterified to glycerol, found in small amounts in butter.
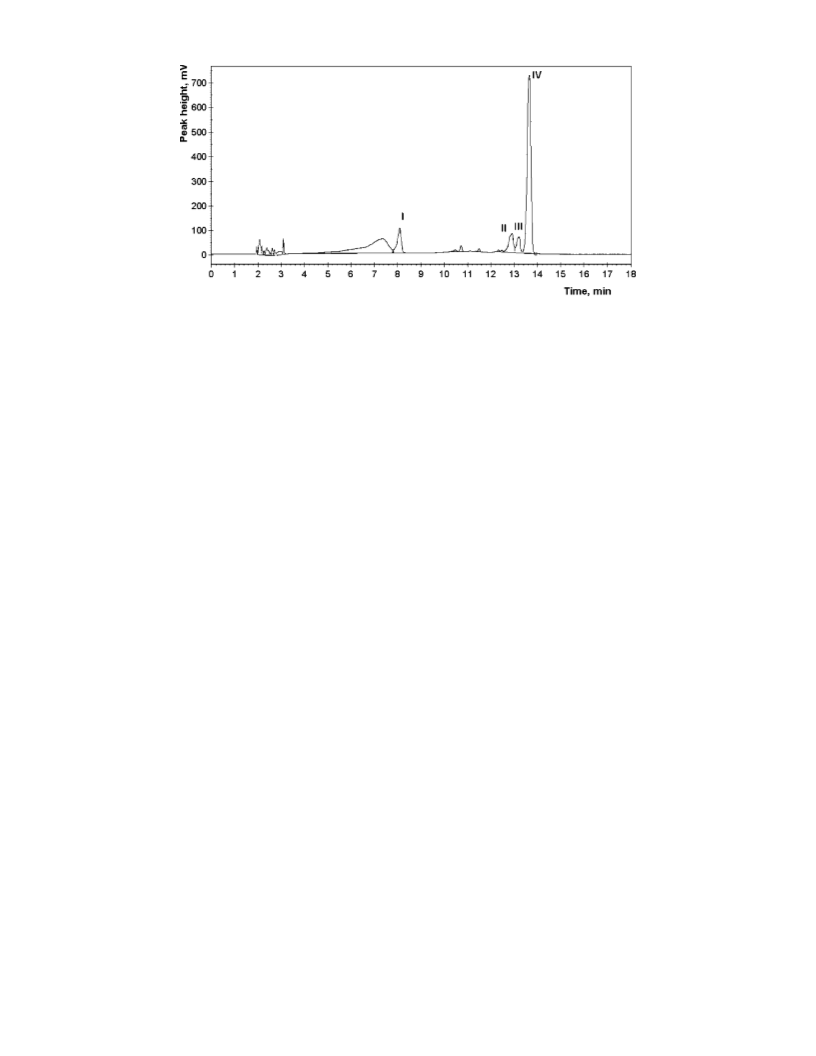
Quantification of Lycopene Isomers. The lycopene isomers were
quantified by analytical HPLC, using a dedicated 150 × 4.6 mm i.d.,
3
µm, Develosil RP-Aqueous C30 column (Phenomenex Inc., Torrance,
CA), protected by a C18 SecurityGuard column, both kept at 25 °C. A
The experiment used a solution of 13-cis-lycopene in tributyrin
(concentration of 19.8 mg/L) and a solution of all-trans-lycopene in
tributyrin (concentration of 20.5 mg/L). A 1-mL aliquot of isomer
solution was mixed with 2 mL of simulated gastric juice (composi-
tion: 0.7% hydrochloric acid, 0.2% sodium chloride, 1.07 ppm pepsin
in distilled water; pH 1.5; Fisher Scientific, Fairlawn, NJ) and 5 µL of
Tween-20. The emulsion was prepared by sonication on ice in an
XL2020 instrument (Heat Systems Inc., Farmingdale, NY) set at level
5, using six 10-s steps alternating with 10-s rest. The simulated gastric
juice (SGJ) was preheated at 37 °C, and the temperature of the emul-
sion after preparation did not exceed 42 °C in any of the samples.
An aliquot of the emulsion was stored at room temperature to check
the stability and 48 h after preparation did not show any phase
separation.
50-µL sample of material was injected, using a loop of 50 µL (Rheodyne
L. P., Rohnert Park, CA). The HPLC method used to quantify lycopene
isomers was derived from the method reported by Yeum et al. (14),
modified to improve resolution while decreasing the resolution time.
Solvent A was a mixture of methanol:MTBE:water (83:15:2, v/v/v),
and solvent B was methanol:MTBE:water (18:80:2, v/v/v). The method
duration was 18 min, at a flow rate of 1 mL/min. The solvent delivery
scheme consisted of a linear change from 90% solvent A + 10% solvent
B at 0 min to 5% solvent A + 95% solvent B at 4 min, followed by
a flat profile for the next 12 min and then a linear change back to 90%
solvent A + 10% solvent B at 18min. â-8′-Apocarotenal was used as
internal standard (15), both for building the calibration curve and in
the experimental injections.

 Moraru, Catalin
Moraru, Catalin