H.M. Badawi et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 152 (2016) 92–100
99
A strong Raman line is observed at 1457 wavenumbers that is
calculated to be highly mixed and consisting mostly of methyl
deformations in both the ethyl groups and of m-CH in plane bend.
At 1431 and 1388 cmÀ1 appear two further strong Raman lines,
which are mainly consisting of rocking deformations of the methyl
and methylene groups in the two ethyl subunits, of methylene
rocking, and of the rocking deformation of the NC2H+ group
(Table 4). At 1272 cmÀ1 a very strong Raman line is seen which
is predicted to be highly mixed and to have 23% ring breathing.
The next lower features in both spectra are of weak or at most of
medium intensity (Fig. 2). The strong intensity Raman lines
observed at 1094 and 1044 cmÀ1 are composed of ring stretching
deformation, m-CH in plane bending, and rocking deformations
of the two methyl groups at the ring (Table 4). The next lower
strong spectral feature is observed in the infrared spectrum at
788 cmÀ1 and consists of m-CH (47%) and p-CH (46%) wagging
vibrations. Then follows at lower wavenumbers, again a series of
weak, very weak, or medium intensity spectral features.
In the Raman spectrum of the neutral lidocaine in the region
around 600–700 cmÀ1 there are two remarkable very strong lines,
one at 708 cmÀ1, consisting of HNC wagging and HNC twisting,
another is at 619 cmÀ1, consisting of NCO wagging and ring-N in
plane bending [14]. In the Raman spectrum of the monohydrate
salt most lines are very weak or weak, medium at most, with the
lines containing NCO scissoring, ring torsional deformation or even
deformation of the hydrate water (Fig. 2). The HNC twisting mode
can be found at 560 cmÀ1 (71% PED) with weak intensity and the
HNC wagging is at 337 cmÀ1 (61% PED) with even very weak inten-
sity in the Raman spectrum of the salt (Table 4). The ring-N in
plane bend cannot be seen at all in the monohydrate Raman
spectrum.
deviation (RMSD) between experiment and theory for the H atoms
chemical shifts is calculated to be 2.32 ppm (Table 3). The RMSD
for the C atoms chemical shifts is calculated to be 8.21 ppm. The
largest experimental-calculated difference in the chemical shift
(d) is about 9.76 ppm for carbon atom C3 of the benzene ring.
For the methyl carbon atoms (C18 and C21) attached to the aro-
matic ring, the difference is about 2 and 4 ppm. For the methylene
(C13 and C14) and methyl carbons (C15 and C16) of the N-ethyl
moiety the difference is found to be in the range 2–5 ppm (Table 3).
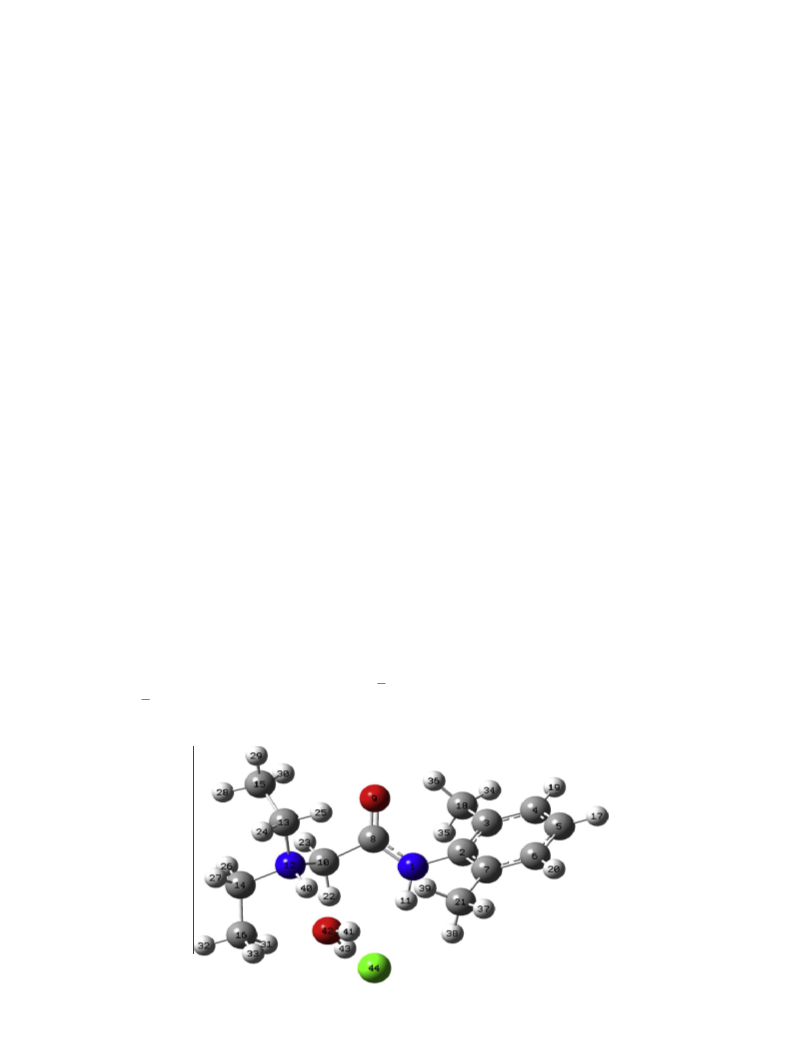
The 1H NMR spectrum of anhydrous lidocaine hydrochloride
matches with its hydrate except that the latter has the water signal
appearing at a chemical shift (d) of 2.06 ppm. Usually, a water sig-
nal is known to be concentration dependent, however, for the cur-
rent hydrate the position of the signal remained the same at
various concentrations of the salt in deuterochloroform. The pro-
tons (H28–H33) of the two methyl groups in N(CH2CH3)2 appeared
as a six-proton triplet at d 1.50 ppm. It is interesting to note that
the CH2 protons in N(CH2CH3)2 (H24–27) become magnetically
nonequivalent as a result of the presence of the chiral center at
the protonated N(12); the diasterotopic protons (H24 and H25 or
H26 and H27) appeared as separate signals at d 3.40 ppm as a dou-
blet of quartet of doublet (dqd) and at d 3.54 ppm as a quartet of
doublet (qd). The splitting pattern in dqd suggests the participation
of the N(12)ÀH(40) bond coupling as also confirmed by the split-
ting of neighboring protons (H22, H23) into a doublet at d
4.42 ppm (Fig. 3). The H11 and H40 are exchangeable protons; in
the 1H NMR spectrum, taken after the sample was treated with
D2O, revealed the absences of the corresponding signals at d
10.18 and 11.07 ppm, respectively. The singlet signal at d 2.25 is
attributed to the two CH3 protons marked H34 to 39. While aro-
matic proton H17 appeared at d 7.06 ppm, aromatic protons
marked H19 and H20 had absorptions at d 7.02 ppm. Restricted
rotation around the crowded C(2)ÀN(1) bond makes H19 and
H20 somewhat different as suggested by the splitting of the neigh-
boring H17 as a doublet of doublet instead of a triplet. For the H
atoms chemical shifts, the largest experimental-calculated differ-
ence of d 6.36 ppm for H43 of H2O is understandable since the loca-
tions of hydroxyl protons are difficult to predict.
The medium intensity band at 596 cmÀ1 in the infrared spec-
trum of the hydrochloride monohydrate is composed of H2O defor-
mation and naturally not present in the neutral compound. In the
infrared spectrum of the parent compound there is a strong band at
598 cmÀ1 which is composed of mainly ring deformations [14]. The
very weak shoulder in the Raman spectrum of the hydrochloride
monohydrate at 382 cmÀ1 is composed mainly of NC2H symmetric
deformation and methylene wagging deformations. In the same
spectrum at 542 cmÀ1 of the parent compound there is a medium
band composed of ring deformations [14]. At 528 cmÀ1 in the
hydrated salt there is a weak band in the infrared spectrum which
is composed mainly of HNC twisting deformation (Table 4). A weak
band in the parent at 523 cmÀ1 is again ring deformation [14]. Thus
there are three distinct bands in the spectrum of the parent
compound, but only a medium one together with a weak and very
weak one in the spectrum of the hydrate. The presence of the water
molecule and the chloride ion changes the geometry and therefore
the observed and calculated line intensities and positions.
In this region also the last strong line at 489 wavenumbers
appears in the infrared spectrum of the parent which is composed
of ring bending deformation and NCO rocking deformation [14]. In
the infrared spectrum of the hydrated salt there is no more strong
intensity band down to 420 wavenumbers, where the last weak
band is seen in the infrared spectrum. The lowest line in the
Raman spectrum of the salt is observed at 242 cmÀ1 with strong
intensity. It is calculated to have 82% methyl torsions in the two
ethyl subgroups.
Lidocaine hydrochloride monohydrate has nine different carbon
signals as expected. The signal at d 164.15 is assigned to the car-
bonyl signals (C8). The aromatic carbons marked C2–7 appeared
in the range d 127.48 to 135.11 ppm. The NMR signals of C10, 13,
15, and 18 are displayed at d 50.94, 49.13, 10.00, and 18.69 ppm,
respectively (Fig. 4).
On comparison with the neutral lidocaine, the effect of the differ-
ence between the neutral nitrogen in lidocaine and its protonated
form in lodicaineÁHClÁH2O salt is expected to cause differences in
the chemical shifts of the magnetic nuclei in the vicinity of the nitro-
gen centers. As such, the 1H spectrum reveals that the protons (Hs)
marked 22/23, 24/25 and 28/29/30 of lidocaineÁHClÁH2O (Fig. 3) are
shifted downfield (i.e. to higher values of chemical shifts d) com-
pared to the corresponding protons of lidocaine (Fig. 3). The down-
field shifts are due to the greater electronegativity of NH+ compared
to N; the inductive withdrawal of the r-electrons reduces the neg-
ative charge densities of the Hs. The magnitude of the decrease in
the charge density and the chemical shifts is the most for the nearest
neighbors. The situation for the 13C chemical shifts, however, is
somewhat complex. The protonation also leads to the change in
13C chemical shifts
been defined as a shift induced by protonation [32,33]. The defini-
tion assumes that the protonated forms exhibit no stereochemical
change with respect to the free base. However, since there are some
D
dC (i.e. dC protonated form À dC free base), that has
4.3. 1H and 13C NMR spectral assignments
The spectral assignments were made based on substituent
effects and multiplicity. The experimental chemical shifts were
compared to the DFT calculated ones in Table 3. On comparison,
the calculated and observed chemical shifts of lidocaineÁHClÁH2O
are in excellent agreement (Table 3). The root mean square
changes in the stereochemistry,
DdC may be considered as the com-
bined effects of both protonation and geometry change. Contrary to
the changes in proton chemical shifts, protonation causes upfield
shifts for most of the 13C signals [33].

 Badawi, Hassan M.
Badawi, Hassan M.