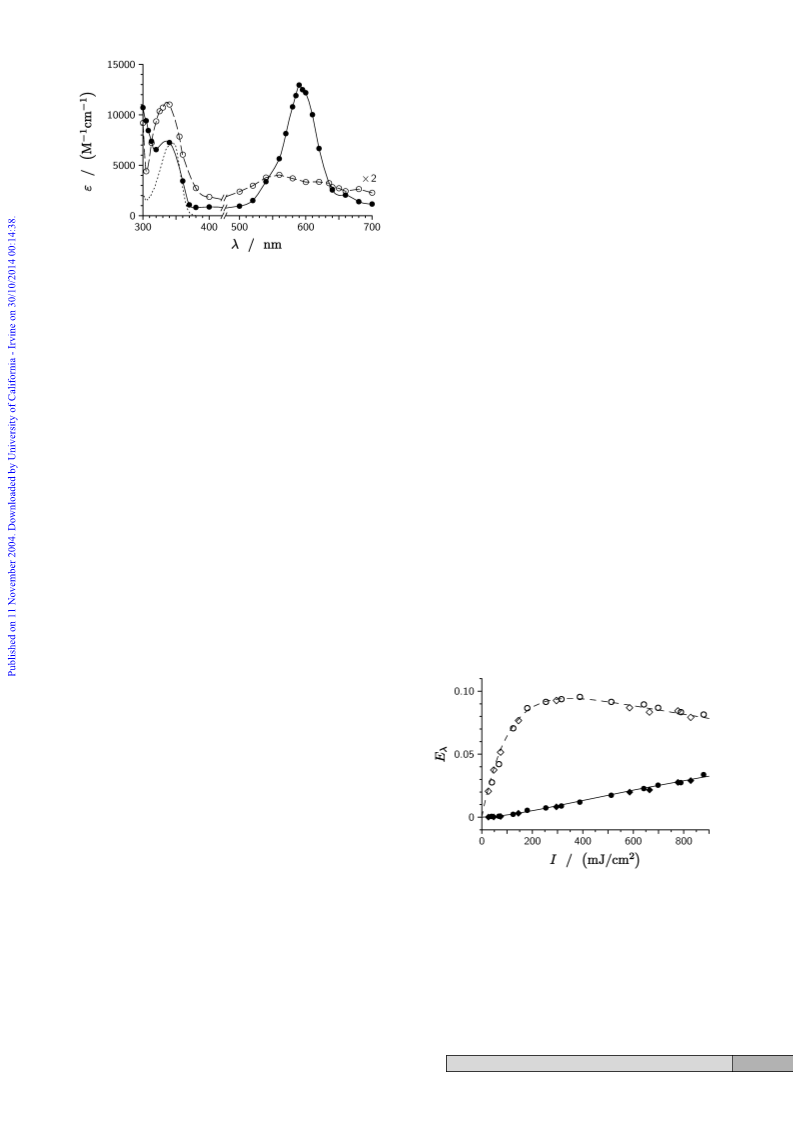
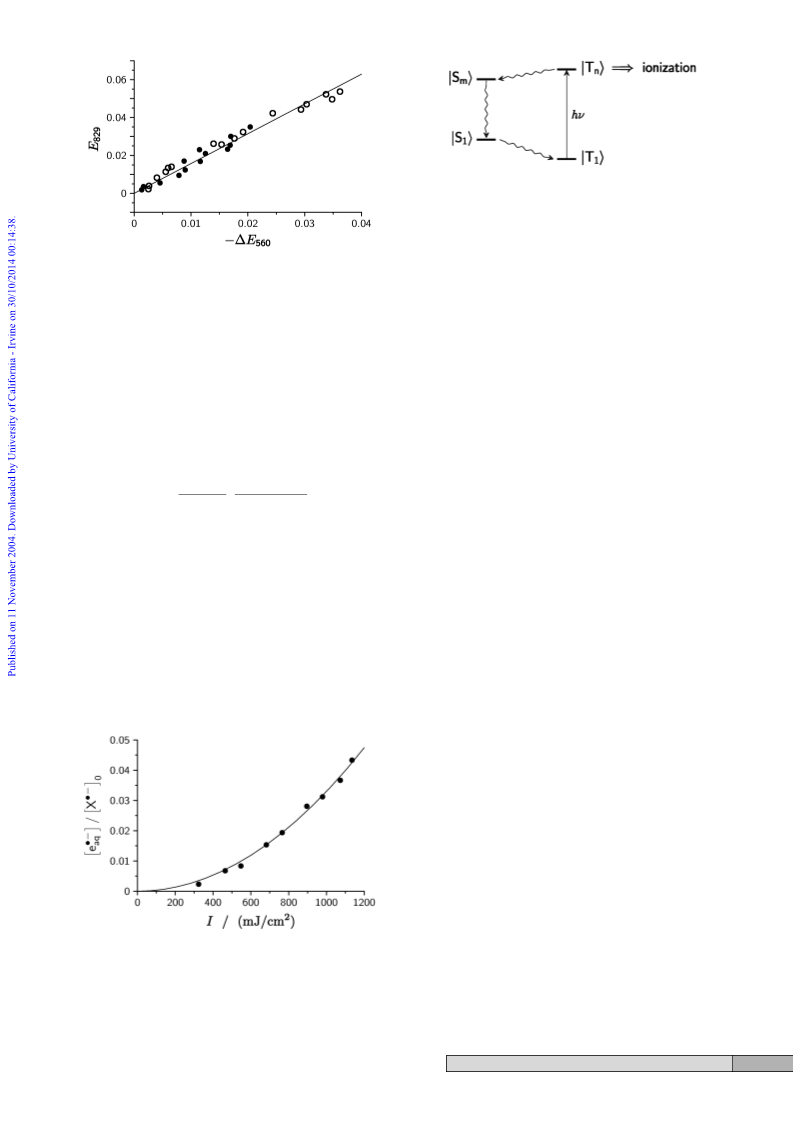
former can interact with the n orbital (b
chromophore but the latter, as well as the carbonyl p and p*
orbitals (b ), remain isolated. While pp* and np* remain the
configurations of the two lowest excited singlet and triplet
1
) of the carbonyl
Absolute concentrations of the transients can be reliably
determined because of the well-defined geometrical arrange-
ment. By a flow-through system it is ensured that each laser
flash hits fresh solution. Detection of the absorbances is
achieved with a high pressure xenon lamp, filters to suppress
stray light, grating monochromator, and a photomultiplier
with an increased sensitivity in the red (Hamamatsu R 928).
The rise time of the detection system is 5 ns, which is essentially
determined by the preamplifier. Signals acquisition and aver-
aging are done with a digital storage oscilloscope coupled to a
personal computer.
2
1
2
states, upper excited states will also include excitations invol-
ving an a2 ring orbital, i.e., also be of symmetry B1 or B2.
Intersystem crossing between any pair of excited configurations
is symmetry-allowed provided that they do not belong to the
same irreducible representation. Hence, the number of potential
intersystem crossing pathways quickly rises for upper excited
states; in addition, the Franck–Condon factors will become
more favorable because within each multiplicity the density of
states increases, and the exchange interaction causes singlet and
triplet functions to be interspersed. Very fast deactivation of the
upper excited triplet states by reverse intersystem crossing
followed by radiationless decay and intersystem crossing back
All chemicals were obtained commercially in the highest
available purity and used as received. The solvent was ultra-
ꢁ1
pure Millipore MilliQ water (resistance 18.2 MO cm ). The
pH was adjusted with KOH. Oxygen was removed by purging
the solutions with argon or N O, and all experiments were
2
to |T
1
i, as shown in Scheme 5, thus seems very likely.
carried out in an inert atmosphere.
While in principle an analogous pathway would be concei-
vable for an excited doublet via the quartet manifold, it is
inaccessible for the two lowest excited states of the radical
anion: The associated doublet–doublet transitions must be
from the HOMO to the SOMO and from the SOMO to the
LUMO, and the resulting configurations are by necessity
doublets; a quartet state can only arise when an electron is
promoted from the HOMO to the LUMO, i.e., at still higher
excitation energies.
Experimental corroboration is provided by luminescence
measurements. In water, xanthone exhibits a weak fluorescence
from a higher singlet state when it is excited in the near UV.
In our photoionizations via the triplet state, the fluorescence
spectrum remained unchanged but the emission intensity
strongly increased with increasing excitation intensity. The
latter effect was absent in our photoionizations via the radical
anion, which is impossible to explain by quenching of the
upper excited state because the emitting state is so short-lived
The extinction coefficient of the electron was measured
relative to its maximum in a difference experiment (with and
without N O); the maximum value was taken from ref. 34.
2
References
1
2
3
Y. Hirata and N. Mataga, Prog. React. Kinet., 1993, 18, 273–308.
H. Gorner, J. Photochem. Photobiol. B, 1994, 26, 117–139.
S. Steenken and L. Goldbergerova, J. Am. Chem. Soc., 1998, 120,
3928–3934.
¨
3
3
4
G. A. Papadantonakis, R. Tranter, K. Brezinsky, Y. Yang, R. B.
van Breemen and P. R. LeBreton, J. Phys. Chem. B, 2002, 106,
7
704–7712.
P. Natarajan and R. W. Fessenden, J. Phys. Chem., 1989, 93, 6095
6100.
5
6
7
8
–
N. Ishiwata, H. Murai and K. Kuwata, J. Phys. Chem., 1993, 97,
7129–7131.
P. Jacques, X. Allonas, A. Sarbach, E. Haselbach and E. Vauthey,
Chem. Phys. Lett., 2003, 378, 185–191.
M. Goez and V. Zubarev, Angew. Chem. Int. Ed. Engl., 1997, 36,
3
3
(
o1 ns) that it cannot be appreciably quenched at our donor
concentrations. These observations indicate that an enhanced
population of upper excited singlet states occurs in xanthone
photoionizations via the triplet but not in those via the radical
anion. Unfortunately, a quantitative investigation is impossible
with our present setup because the detected signal is a con-
volution integral of the rapidly varying luminescence during
the laser pulse with the much slower response function of our
detection system.
2
664–2666.
9
0
M. Goez and V. Zubarev, J. Phys. Chem. A, 1999, 103, 9605–9613.
M. Goez, V. Zubarev and G. Eckert, J. Am. Chem. Soc., 1998,
120, 5347–5348.
1
11 M. Goez and V. Zubarev, Chem. Phys., 2000, 256, 107–116.
1
2
J. J. Cavaleri, K. Prater and R. M. Bowman, Chem. Phys. Lett.,
996, 259, 495–502.
J. C. Scaiano, J. Am. Chem. Soc., 1980, 102, 7747–7753.
1
1
1
3
4
F. Wilkinson and A. Garner, J. Chem. Soc., Faraday Trans. 2,
Taking into account the apparently intrinsically more favor-
able photoionization efficiency of radical anions, the increase
of the electron yield by the catalytic cycle (Scheme 2), the much
longer life of radical anions compared to excited states when
the access of bimolecular reaction partners is restricted
1
15 K. Abdullah and T. J. Kemp, J. Chem. Soc., Perkin Trans. 2, 1985,
977, 73, 222–233.
1279–1283.
M. Kaise and K. Someno, Chem. Lett., 1987, 1295–1298.
1
1
6
7
M. Goez and I. Sartorius, J. Am. Chem. Soc., 1993, 115, 11123–
1
1133.
(
e.g., when the radical anions are immobilized in a membrane),
1
8
S. Sekiguchi, Y. Kobori, K. Akiyama and S. Tero-Kubota, J. Am.
Chem. Soc., 1998, 120, 1325–1326.
19 C. Hoegemann and E. Vauthey, J. Phys. Chem. A, 1998, 102,
and the fact that substrates (e.g., ketones or quinones) and
quenchers (e.g., amines or amino acids) that are typical for
the mechanism of Scheme 2 are ubiquituous in nature, it
does not seem inconceivable that such photoionizations via
radical anions might also play a role in biologically relevant
systems.
10051–10059.
2
2
2
2
2
2
0
1
2
3
4
5
G. T. Davis, M. M. Demek and D. H. Rosenblatt, J. Am. Chem.
Soc., 1972, 94, 3321–3325.
D. A. Dunn, D. I. Schuster and R. Bonneau, J. Am. Chem. Soc.,
1
985, 107, 2802–2804.
A. M. Halpern, D. A. Forsyth and M. Nosowitz, J. Phys. Chem.,
986, 90, 2677–2679.
1
Experimental section
M. S. Workentin, L. J. Johnston, D. D. M. Wayner and V. D.
Parker, J. Am. Chem. Soc., 1994, 116, 8279–8287.
J. W. T. Spinks and R. J. Woods, An Introduction to Radiation
Chemistry, Wiley, New York, 1976.
The samples were excited with an excimer laser (Lambda
Physik LPX-210i, 308 nm, effective pulse duration 60–80 ns
owing to an afterpulse) and/or an Nd:YAG laser (Continuum
Surelite-II, 532 or 355 nm, pulse width below 10 ns). In two-
pulse experiments, arbitrary positive or negative interpulse
delays can be selected with a home-made trigger generator.
The two laser beams are set up as to enter the sample in a
collinear geometry, and to provide homogeneous illumination
of the observed volume (2 mm in the direction of the exciting
beams, 4 mm in that of the monitoring beam, and 2 mm
perpendicular to the other two) with high photon fluxes.
R. Boch, M. K. Whittlesey and J. C. Scaiano, J. Phys. Chem.,
1994, 98, 7854–7857.
26 M. W. Ferguson, P. C. Beaumont, S. E. Jones, S. Navaratnam and
B. J. Parsons, Phys. Chem. Chem. Phys., 1999, 1, 261–268.
2
7
8
J. K. Hurley, H. Linschitz and A. Treinin, J. Phys. Chem., 1988,
2, 5151–5159.
H. Lutz, E. Bre
758–1762.
9
2
heret and L. Lindqvist, J. Phys. Chem., 1973, 77,
´ ´
1
29 T. Shida, S. Iwata and M. Imamura, J. Phys. Chem., 1974, 78,
741–748.
5
496
P h y s . C h e m . C h e m . P h y s . , 2 0 0 4 , 6 , 5 4 9 0 – 5 4 9 7
T h i s j o u r n a l i s & T h e O w n e r S o c i e t i e s 2 0 0 4

 Goez, Martin
Goez, Martin