8454 J . Org. Chem., Vol. 63, No. 23, 1998
Smith et al.
thorough safety testing on any procedure under consid-
eration. We urge great care in scaling up any nitration
procedure.
under reduced pressure (30 mmHg), and a second, lower-
pressure distillation (0.2 mmHg) at temperatures up to
about 70 °C was used to provide the products, leaving
the catalyst behind in the reaction flask. The composi-
tion of the distilled product mixture was determined by
both GC and NMR techniques, and where possible, the
spectra were compared with those of authentic samples.
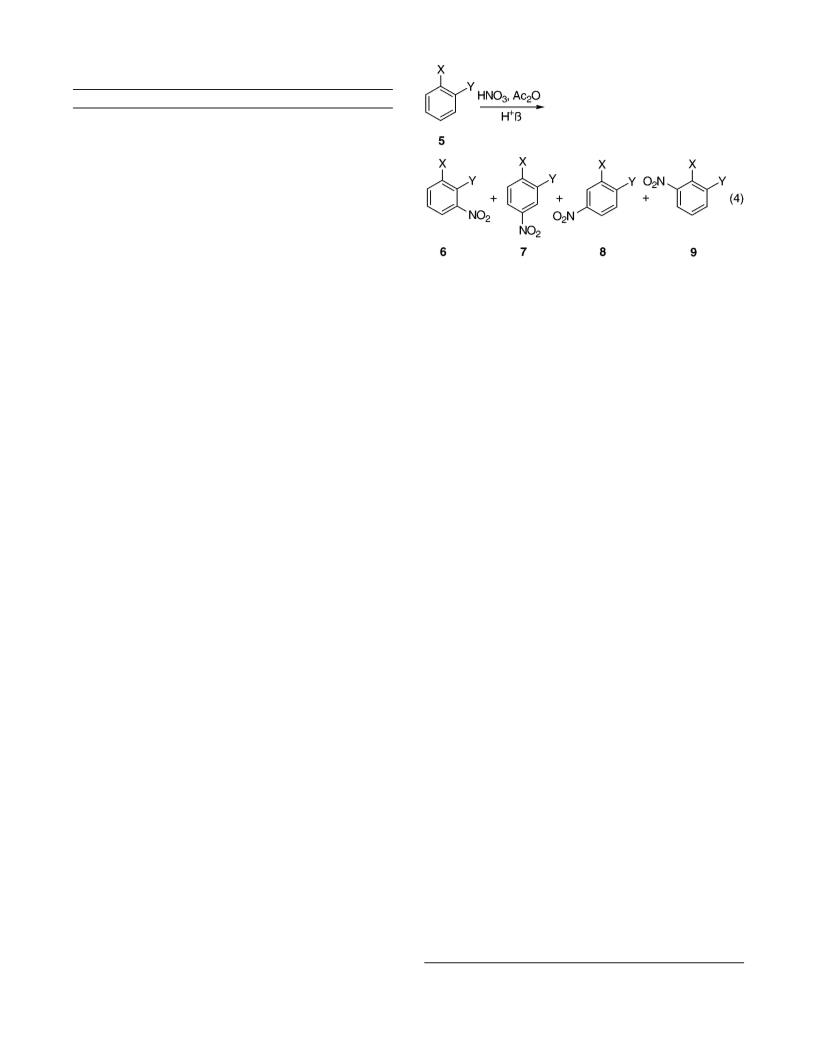
Yields and isomer proportions are listed in Tables 8 and
9. Authentic samples were available for comparison with
the products from the nitration of monosubstituted
benzenes. NMR data for individual products from di-
substituted benzenes are given below.
Con clu sion
Zeolite H+â is an effective catalyst for the nitration of
monosubstituted aromatic substrates by acetic anhydride-
nitric acid mixtures. Benzene, alkylbenzenes, and ha-
logenobenzenes are nitrated in quantitative yields with
excellent para selectivities using a stoichiometric quan-
tity of nitric acid and enough acetic anhydride to convert
all the nitric acid into acetyl nitrate and all the water
into acetic acid. Dinitro compounds are not formed in
any significant amounts, and the best para regioselection
is obtained, at least in the case of toluene, by the addition
of the reagents in the order nitric acid/zeolite, acetic
anhydride, substrate. Under such conditions, the para
regioselectivities obtained from a range of substrates
(Table 8) are generally the best ever obtained in high-
yielding reactions. Furthermore, the method has a
number of practical advantages (no solvent; moderate
temperature; easy separation by direct vacuum distilla-
tion of both the only byproduct, acetic acid, and the
reaction product; and easy recycling of the catalyst) that
should make it highly attractive for commercial applica-
tion. Patent protection has therefore been sought,9 and
the safety of the process has been considered in detail.
The method also offers advantageous regioselectivity
compared with traditional methods for nitration of some
1,2-disubstituted benzenes (Table 9).
F r om Nitr a tion of 1,2-Diflu or oben zen e (5a ). 1,2-
Difluoro-4-nitrobenzene (7a ): 1H NMR (CDCl3, 400 MHz)
δ 7.42 (1H, m), 8.12 (2H, m); 19F NMR (CDCl3, 400 MHz)
δ -126.7 (1F, m), -133.1 (1F, m).
F r om Nitr a tion of 2-Ch lor oflu or oben zen e (5b).
2-Chloro-1-fluoro-4-nitrobenzene (8b): 1H NMR (CDCl3,
400 MHz) δ 7.34 (1H, m), 8.21 (2H, m), 8.35 (1H, m); 19
F
NMR (CDCl3, 400 MHz) δ -104.40. 1-Chloro-2-fluoro-
4-nitrobenzene (7b): 1H NMR (CDCl3, 400 MHz) δ 7.63
(1H, m), 8.04 (2H, m); 19F NMR (CDCl3, 400 MHz) δ
-110.31.
F r om Nitr a tion of 2-Br om oflu or oben zen e (5c).
2-Bromo-1-fluoro-4-nitrobenzene (8c): 1H NMR (CDCl3,
400 MHz) δ 7.29 (1H, m), 8.22 (1H, m), 8.46 (1H, m).
1-Bromo-2-fluoro-4-nitrobenzene (7c): 1H NMR (CDCl3,
400 MHz) δ 7.77 (1H, m), 7.95 (2H, m).
F r om Nitr a tion of 2-F lu or oa n isole (5d ). 2-Fluoro-
1
4-nitroanisole (7d ): H NMR (CDCl3, 400 MHz) δ 4.01
(3H, s), 7.05 (1H, m), 8.00 (1H, m), 8.09 (1H, m); 19F NMR
(CDCl3, 400 MHz) δ -131.38. 2-Fluoro-6-nitroanisole
(9d ): 1H NMR (CDCl3, 400 MHz) δ 4.07 (3H, s), 7.07 (1H,
m), 7.38 (1H, m), 7.60 (1H, m); 19F NMR (CDCl3, 400
MHz) δ -126.70.
Exp er im en ta l Section
P r ep a r a tion of th e P r oton F or m of Zeolite â. The
zeolite (CP806), as supplied by PQ Zeolites Ltd., was
calcined at 600 °C for 12 h to remove the organic
template. The zeolite was then stirred in a refluxing
solution (1 M) of aqueous ammonium acetate (10 mL/g)
for 1 h. After filtration and a second exchange, to ensure
complete ion exchange, the zeolite was again filtered and
then calcined at 600 °C overnight, cooled, and ground to
a fine powder. It was reheated to 400 °C for 2 h
immediately prior to use.
Nitr a tion P r oced u r e: In vestiga tive Exp er im en ts.
Nitric acid (0.25 g, 70%, 2.5 mmol), acetic anhydride (20
mL), 3A molecular sieves (0.2 g), zeolite (0.1 g), toluene
(0.23 g, 2.5 mmol), and hexadecane (0.3 g, 1.3 mmol) as
internal standard were mixed together in the desired
order and stirred at room temperature. The reaction was
sampled at regular intervals and analyzed by GC. Later
it was found that the excess of acetic anhydride could be
reduced and the molecular sieves omitted from the
mixture with only minor modifications to the procedure.
Such a procedure was therefore adopted for preparative
experiments (see below).
Nitr a tion P r oced u r e: P r ep a r a tive Exp er im en ts
(N.B.: See Section on Sa fety). Nitric acid (2.5 g, 35
mmol) was mixed with H+â (1.0 g) and stirred in an ice-
water bath for a few minutes. Stirring and cooling were
maintained while acetic anhydride (5.0 mL, 53 mmol)
was added (the temperature rose temporarily to about
12 °C during this process), followed by the substrate (35
mmol), dropwise. The cooling bath was removed, and the
mixture was stirred for 30 min unless stated otherwise
in the text. The acetic acid was removed by distillation
F r om Nitr a tion of 2-F lu or otolu en e (5e). 2-Fluoro-
5-nitrotoluene (8e): 1H NMR (CDCl3, 400 MHz) δ 2.20
(3H, s), 7.39 (1H, m), 7.88 (1H, m), 7.98 (1H, m); 19F NMR
(CDCl3, 400 MHz) δ -113.40. 2-Fluoro-4-nitrotoluene
(7e): 1H NMR (CDCl3, 400 MHz) δ 2.38 (3H, s), 7.15 (1H,
m), 8.09 (2H, m); 19F NMR (CDCl3, 400 MHz) δ -106.50.
F r om Nitr a tion of 2-Ch lor otolu en e (5f). 2-Chloro-
5-nitrotoluene (8f): 1H NMR (CDCl3, 400 MHz) δ 7.41
(1H, d, J 8.4), 7.99 (1H, dd, J 8.4, 2.3), 8.15 (1H, d, J
2.3). 2-Chloro-4-nitrotoluene (7f): 1H NMR (CDCl3, 400
MHz) δ 7.48 (1H, d, J 8.7), 7.07 (1H, dd J 2.4, 8.7), 8.08
(1H, d, J 2.6).
F r om Nitr a tion of 2-Br om otolu en e (5g). 2-Bromo-
5-nitrotoluene (8g): 1H NMR (CDCl3, 400 MHz) δ 7.39
(1H, d, J 8.4), 8.03 (1H, dd, J 8.4, 2.3), 8.31 (1H, d, J
2.3). 2-Bromo-4-nitrotoluene (7g): 1H NMR (CDCl3, 400
MHz) δ 7.66 (1H, d, J 8.7), 7.86 (1H, dd J 2.6, 8.7), 8.05
(1H, d, J 2.7).
Ack n ow led gm en t. A.M. thanks ZENECA for finan-
cial support. We also thank the Engineering and Physi-
cal Sciences Research Council (EPSRC) and the Uni-
versity of Wales for the grants which enabled purchase
of the NMR equipment used in this study and the
EPSRC mass spectrometry service in Swansea for
running mass spectra. Gifts of zeolites from PQ Zeolites
(now Zeolyst International) are gratefully acknowl-
edged. We also thank Dr. P. G. Urben for useful
discussions concerning safety aspects of nitration
chemistry.
J O981557O

 Smith, Keith
Smith, Keith