CHEMOENZYMATIC SYNTHESIS
1141
by vigorously stirring for 30 min. To this preparation was to afford the product as a white semicrystalline waxy mater-
added stearic acid (50.6 g; 178 mmol), immobilized R. miehei ial (41.1 g; 94% yield). It was possible to prepare crystalline
lipase (3.5 g), and molecular sieves (10 g). The mixture was material for melting point determination using ethyl acetate
suspended in diethyl ether (200 mL) and stirred on a magnetic at −18°C. M.p. = 33.5–34.0°C. 1H NMR δ 5.40–5.33
stirrer hot-plate at room temperature for 48 h. The reaction (m, 10H, =C–H), 5.34–5.25 (m, 1H, –CH2–CH–CH2), 4.29
was continuously monitored by analytical TLC (silica gel; (dd, J = 11.9 Hz, J = 4.3 Hz, 2H, –CH2–CH–CH2–), 4.13
eluted with 80:20:2 petroleum ether/diethyl ether/acetic acid). (dd, J = 11.9 Hz, J = 5.9 Hz, 2H, –CH2–CH–CH2–),
The lipase and silica gel were separated off by filtration, and 2.85–2.77 (m, 8H, =C–CH2–C=), 2.35–2.26 (2 × t, J = 7.6 Hz,
the solvent was removed in vacuo. Recrystallization of the re- 6H, –CH2–COO–), 2.11–2.03 (m, 4H, =CH–CH2–CH2–
sulting solid material in methanol afforded the product as and CH3–CH2–CH=), 1.72–1.65 (m, 2H, –CH2–CH2–COO–
highly pure fine-powdered white crystals (82.3 g; 74% yield). in EPA), 1.62–1.56 (m, 4H, –CH2CH2COO– in stearic acid),
M.p. = 77.8–78.4°C. 1H NMR δ 4.21–4.08 (m, 5H, 1.40–1.16 (m, 56H, –CH2–), 0.96 (t, J = 7.5 Hz, 3H, –CH3
–CH2–CH–CH2–), 2.51 (br s, 1H, –OH), 2.35 (t, J = 7.4 Hz, in EPA), and 0.87 (t, J = 6.4 Hz, 6H, –CH3) ppm. 13C δ 173.2
4H, –CH2COO–), 1.62 (m, 4H, –CH2–CH2–COO–), (2; α C=O), 172.5 (1; β C=O), 131.9 (1), 128.9 (1), 128.7
1.40–1.16 (m, 56H, –CH2–), and 0.87 (t, J = 6.3 Hz, 6H, (1), 128.5 (1), 128.2 (1), 128.1 (2), 128.0 (1), 127.8 (1), 126.9
–CH3) ppm. 13C δ 173.9 (2; α C=O), 68.3 (1), 65.0 (2), 34.1 (1), 68.9 (1), 62.0 (2), 34.0 (2), 33.5 (1), 31.9 (2), 29.7
(2), 31.9 (2), 29.7 (10), 29.6 (4), 29.4 (6), 29.2 (2), 29.1 (2), (10), 29.6 (4), 29.4 (4), 29.3 (2), 29.2 (2), 29.1 (2), 26.4 (1),
24.9 (2), 22.7 (2), and 14.1 (2) ppm. IR νmax 3300–3600 (br, 25.6 (3), 25.5 (1), 24.8 (2), 24.7 (1), 22.6 (2), 20.5 (1), 14.2
O–H), 2915 (vs, C–H), 2849 (vs, C–H), 1735 (vs, C=O) (1) and 14.1 (2) ppm. IR νmax 3020 (s, C–H), 2914 (vs, C–H),
cm−1. Elemental analysis: found: C, 74.87; H, 12.30. 2849 (vs, C–H), 1732 (vs, C=O) cm−1. Elemental analysis:
C39H76O5 requires C, 74.94; H, 12.26%.
found: C, 78.05; H, 11.58. C59H104O6 requires C, 77.92; H,
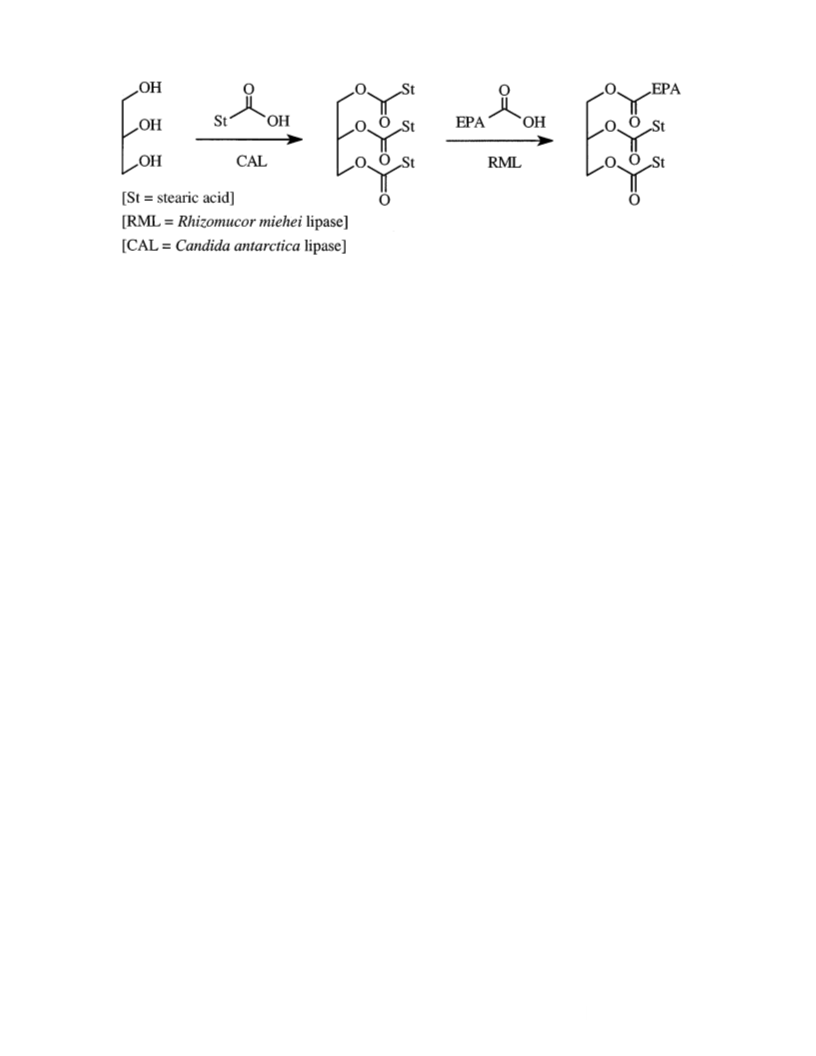
Preparation of tristearin. Immobilized C. antarctica lipase 11.53%.
(1.50 g) was added to a mixture of glycerol (1.55 g, 16.8
Preparation of 2-docosahexaenoyl-1,3-distearylglycerol.
mmol) and stearic acid (15.1 g; 53.1 mmol). The mixture was The procedure was identical to the one for preparing the cor-
gently stirred on a magnetic stirrer hot-plate at 70–75°C under responding 2-EPA-TAG adduct using 1,3-distearin (1.00 g;
continuous vacuum of 0.05–0.1 Torr. The volatile water pro- 1.60 mmol), DHA (0.59 g; 1.80 mmol), DMAP (0.11 g; 0.90
duced during the course of the reaction was continuously con- mmol) and EDCI (0.35 g; 1.80 mmol) in dichloromethane
densed into a liquid nitrogen cooled trap. After 48 h the reac- (8 mL). The product was afforded as a semicrystalline waxy
tion was discontinued, chloroform added, and the enzyme material (1.36 g; 91% yield), but crystals could be afforded
separated off by filtration. Pure product was afforded as a from cold ethyl acetate treatment. M.p. = 35.0–35.5°C.
white crystalline material (13.2 g; 88% yield) after crystal- 1H NMR δ 5.42–5.30 (m, 12H, =C–H), 5.29–5.24 (m, 1H,
lization from chilled chloroform (0–4°C), filtration, and dry- –CH2–CH–CH2), 4.29 (dd, J = 11.9 Hz, J = 4.4 Hz, 2H,
ing in vacuo. M.p. = 69.2–70.0°C. 1H NMR δ 5.34–5.26 (m, –CH2–CH–CH2–), 4.13 (dd, J = 11.9 Hz, J = 5.9 Hz,
1H, –CH2–CH–CH2–), 4.30 (dd, J = 11.9 Hz, J = 4.3 Hz, 2H, 2H, –CH2–CH–CH2–), 2.86–2.77 (m, 10H, =C–CH2–C=),
–CH2–CH–CH2–), 4.14 (dd, J = 11.9 Hz, J = 6.0 Hz, 2H, 2.38–2.35 (m, 4H, –CH2–CH2–COO– in DHA), 2.29 (t,
–CH2–CH–CH2–), 2.31 (t, J = 7.3 Hz, 6H, –CH2–COO), J = 7.4 Hz, 4H, –CH2–COO–), 2.12–2.00 (m, 2H,
1.63–1.58 (m, 6H, –CH2–CH2–COO–), 1.40–1.16 (m, 84H, CH3–CH2–CH=), 1.62–1.54 (m, 4H, –CH2CH2COO– in
–CH2–), and 0.87 (t, J = 6.3 Hz, 9H, –CH3) ppm. 13C NMR δ stearic acid), 1.35–1.18 (m, 56H, –CH2–), 0.96 (t, J = 7.5 Hz,
173.3 (2; α C=O), 172.9 (1; β C=O), 68.8 (1), 62.1 (2), 34.2 3H, –CH3) and 0.86 (t, J = 6.3 Hz, 6H, –CH3) ppm. 13C NMR
(1), 34.0 (2), 31.9 (3), 29.7 (15), 29.5 (6), 29.4 (6), 29.3 (3), δ 173.1 (2; α C=O), 172.0 (1; β C=O) 131.9 (1), 129.4 (1),
29.1 (6), 24.8 (3), 22.7 (3) and 14.1 (3) ppm. IR νmax 2917 128.5 (1), 128.2 (3), 128.0 (2), 127.9 (1), 127.8 (1), 127.5 (1),
(vs, C–H), 2849 (vs, C–H), 1736 (vs, C=O) cm−1. Elemental 126.9 (1), 69.0 (1), 61.9 (2), 34.0 (3), 31.9.(2), 29.7 (10), 29.6
analysis: found: C, 76.89; H, 12.48. C57H110O6 requires C, (6), 29.4 (2), 29.3 (2), 29.2 (2), 29.1 (2), 25.6 (3), 25.5 (1),
76.79; H, 12.44%.
24.8 (3), 22.6 (3), 20.5 (1), 14.2 (1) and 14.1 (2) ppm. IR νmax
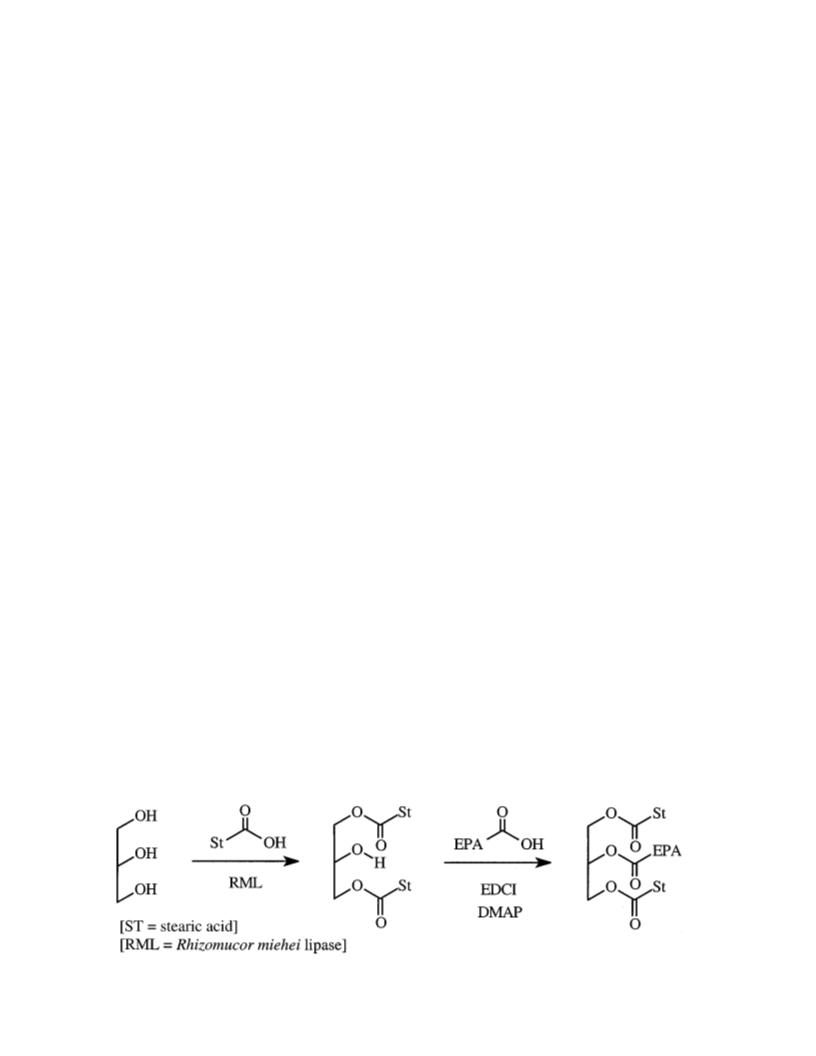
Preparation of 2-eicosapentaenoyl-1,3-distearylglycerol. 3020 (s, C–H), 2914 (vs, C–H), 2849 (vs, C–H), 1733 (vs,
To a solution of 1,3-distearylglycerol (33.0 g; 52.8 mmol) and C=O) cm−1. Elemental analysis: found: C, 78.27; H, 11.45.
EPA as a free acid (16.8 g; 55.4 mmol) in dichloromethane C61H106O6 requires C, 78.32; H, 11.42%.
(300 mL) was added DMAP (3.22 g; 26.4 mmol) and EDCI
Preparation of 1-eicosapentaenoyl-2,3-distearylglycerol.
(12.1 g; 63.0 mmol). The resulting solution was stirred on a Immobilized R. miehei lipase (2.2 g) was added to a mixture
magnetic stirrer hot-plate at room temperature for 24 h. The of tristearin (13.10 g; 14.7 mmol) and EPA as a free acid (8.90
reaction was terminated by passing the reaction solution g; 29.4 mmol). The mixture was gently stirred on a magnetic
through a short column packed with silica gel after reduction stirrer hot-plate at 75°C under continuous vacuum of
of the volume to 50 mL by rotary evaporation in vacuo. Sol- 0.05–0.10 mm Hg for 20 h. The mixture was allowed to cool,
vent removal in vacuo afforded a mixture of the crude prod- chloroform added (25 mL), and the reaction stopped by filter-
uct contaminated with some EPA which was purified by sil- ing off the lipase. The chloroform was removed in vacuo on a
ica gel column chromatography using n-hexane as an eluent rotary evaporator and the residue dissolved in diethyl ether
JAOCS, Vol. 77, no. 11 (2000)

 Haraldsson, Gudmundur G.
Haraldsson, Gudmundur G.