Zampella et al.
The aqueous layer was fractionated by HPLC on the reversed-
CuSO
4
; flow rate, 1.0 mL/min; detection UV, 254 nm; retention
phase Phenomenex Hydro (4 µ, 250 × 4.6 mm) column eluting
times of the standards (min) L-Ser (5.7) and D-Ser (5.1). The
hydrolysate of homophymine A (1) contained D-serine.
2
with MeOH:H O 2:98 (flow rate 0.5 mL/min) to give 2.2 mg of
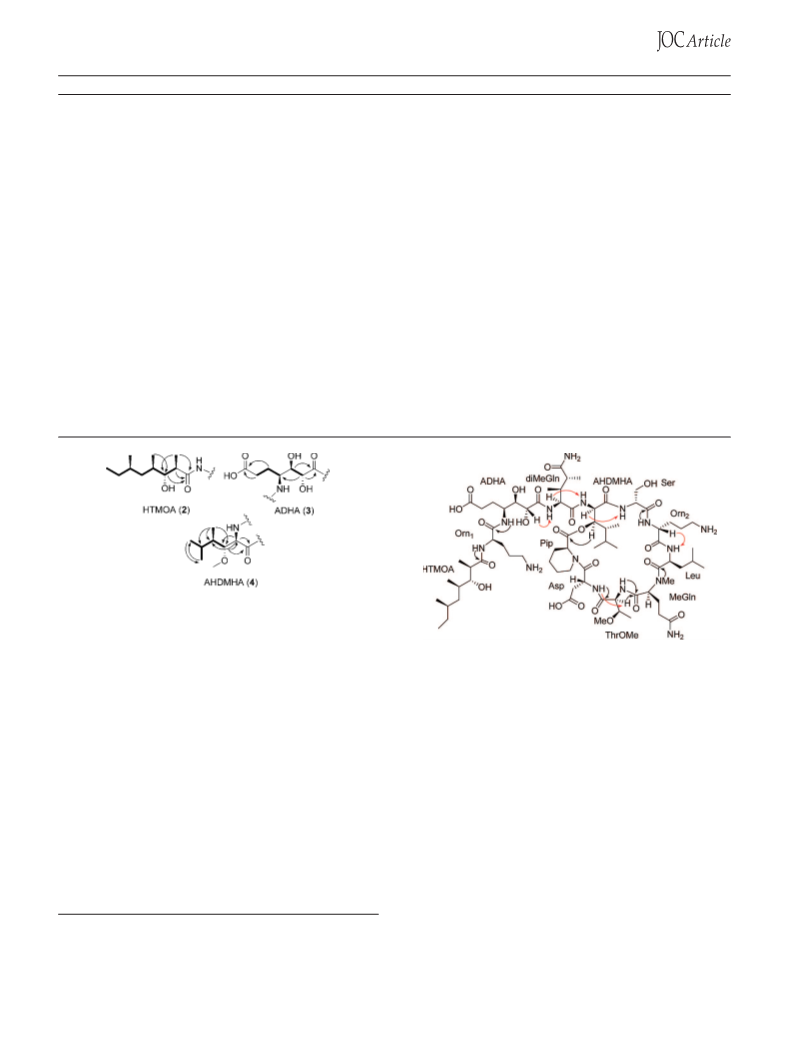
pure 2-amino-3-hydroxy-4,5-dimethylhexanoic acid (4) (R
min). The additional peak at R 3.7 min was further purified by
HPLC on the same column eluting with H O 100% containing
.05% TFA (flow rate 0.5 mL/min) to give 0.7 mg of pure 4-amino-
,3-dihydroxyheptandioic acid (3) (R 34.8 min).
-Amino-2,3-dihydroxyheptandioic Acid (3). ESI/MS m/z
t
40.1
LC-MS Analysis of GITC Derivatives. Triethylamine (10 µL)
and a GITC (2,3,4,6-tetra-O-acetyl-R-D-glucopyranosyl isothiocy-
anate) solution (50 µL, made with 3.9 mg/mL in MeCN) were added
to the acid hydrolysate (100 µg) of 1 or callipeltin A, or an authentic
amino acid standard (100 µg). The reaction mixture was kept for
30 min at room temperature, and then the reaction was quenched
t
2
0
2
t
4
+
1
2
)
08.1 [M + H ]; H NMR (500 MHz, CD
5.2 Hz, H-2), 3.93 (1H, dd, J ) 5.2, 2.0 Hz, H-3), 3.37 (1H, m,
H-4), 2.35 (1H, m, H-6), 2.42 (1H, m, H-6′), 1.91 (2H, m, H-5);
3
OD) δ 4.29 (1H, d, J
2
by adding 40 µL of MeCN-5% AcOH in H O (1:1). An aliquot
was dried under vacuum and then diluted with the same solvent
mixture and subjected to LC-MS analysis as described above, except
for monitoring the absorption at 254 nm. Retention times (min):
L-NMeGlu from callipeltin A (25.3), L-Asp (16.9), D-Asp (17.9),
L-Glu (15.6), and D-Glu (14.5). The hydrolysate of 1 gave peaks
for L-NMeGlu (25.5) and D-Asp (17.9).
1
3
3
C NMR (125 MHz, CD OD) δ 179.4, 171.5, 74.3, 70.5, 53.2,
3
3.5, 27.3.
2
-Amino-3-hydroxy-4,5-dimethylhexanoic Acid (4). ESI/MS
+
1
3
m/z 176.1 [M + H ]; H NMR (500 MHz, CD OD) δ 3.70 (1H, d,
J ) 2.6 Hz, H-2), 3.65 (1H, dd, J ) 10.0, 2.6 Hz, H-3), 2.22 (1H,
m, H-5), 1.84 (1H, m, H-4), 0.94 (3H, d, J ) 7.0 Hz, H-6), 0.85
Stereochemistry of the 4-Amino-2,3-dihydroxyheptandioic
Acid Residue (ADHA, 3). A 1 mg portion of 1 was stirred in a
1
3
(
(
9
3H, d, J ) 6.9 Hz, H-8), 0.80 (3H, d, J ) 7.0 Hz, H-7); C NMR
mixture of EtOH (300 µL) and 1% (w/w) NaIO
for 18 h at ambient temperature. The reaction solution was
concentrated and then dissolved in 700 µL of 50% AcOH/H O and
boiled for 2 h at 102 °C in the presence of 30% H (200 µL)
4
in water (750 µL)
125 MHz, CD
.5.
Determination of the Absolute Configuration: Peptide
3
OD) δ 171.5, 74.0, 59.0, 41.2, 27.2, 21.4, 14.9,
2
2
O
2
Hydrolysis. A peptide sample (2 mg) was dissolved in degassed 6
N HCl (0.5 mL) in an evacuated glass tube and heated at 160 °C
for 16 h. The solvent was removed in vacuo and the resulting
material was subjected to further derivatization.
and concentrated HCl (100 µL). The residue was hydrolyzed,
derivatized with GITC as described above, and analyzed by LC-
MS, which revealed the presence of L-Glu (15.4).
Mosher Analysis of 2. Freshly distilled (+)-R-methoxy-R-
LC-MS Analysis of Marfey’s (FDAA) Derivatives. A portion
(
trifluoromethyl)phenylacetic (MTPA) chloride (6 µL) was added
of the hydrolysate mixture (800 µg) or the amino acid standard
to a solution of 2 (0.5 mg) with a catalytic amount of 4-(dimethy-
(
500 µg) was dissolved in 80 µL of a 2:3 solution of TEA:MeCN
lamino)pyridine and 5 µL of freshly distilled triethylamine, in 1
mL of freshly distilled CH Cl , and the solution was allowed to
2 2
stand at room temperature for 12 h under an argon atmosphere.
The residue, obtained after evaporation of the solvent, was subjected
to reverse-phase HPLC with use of a linear gradient from water
and this solution was then treated with 75 µL of 1% 1-fluoro-2,4-
dinitrophenyl-5-L-alaninamide (FDAA) in 1:2 MeCN:acetone. The
vials were heated at 70 °C for 1 h, and the contents were neutralized
with 0.2 N HCl (50 µL) after cooling to room temperature. An
aliquot of the L-FDAA derivative was dried under vacuum, diluted
(
3
100%) to CH CN (100%), UV detector λ ) 260 nm, to obtain 0.1
2
with MeCN-5% HCOOH in H O (1:1), and separated on a Vydac
mg of the (R)-(+)-MTPA ester of 2. The same procedure was used
C18 (25 × 1.8 mm i.d.) column by means of a linear gradient from
to obtain 0.4 mg of the (S)-(-)-MTPA ester of 2. 3-(+)-(R)-MTPA
1
0
0% to 50% aqueous acetonitrile containing 5% formic acid and
.05% trifluoroacetic acid, over 45 min at 1 mL/min. The RP-HPLC
1
3
ester of 2: selected H NMR (500 MHz, CDCl ) δ 3.68 (1H, dd,
H-3), 3.14 (1H, quintet, H-2), 1.70 (1H, overlapped, H-4), 1.36
system was connected to the electrospray ion source by inserting a
splitter valve and the flow going into the mass spectrometer source
was set at a value of 100 µL/min. Mass spectra were acquired in
positive ion detection mode (m/z interval of 320-900) and the data
were analyzed by using the suite of programs Xcalibur; all masses
were reported as average values. Capillary temperature was set at
(
(
(
(
1
0
3H, d, J ) 6.8 Hz, H-9), 0.92 (3H, d, J ) 7.5 Hz, H-10), 0.89
3H, d, J ) 6.8 Hz, H-11), 0.87 (3H, t, J ) 7.1 Hz, H-8). 3-(-)-
1
3
S)-MTPA ester of 2: selected H NMR (500 MHz, CDCl ) δ 3.68
1H, dd, H-3), 3.12 (1H, quintet, H-2), 1.77 (1H, overlapped, H-4),
.32 (3H, d, J ) 6.7 Hz, H-9), 0.96 (3H, d, J ) 7.1 Hz, H-10),
.91 (3H, d, J ) 6.5 Hz, H-11), 0.87 (3H, t, J ) 7.1 Hz, H-8).
Methanolysis of Homophymine A (1). A solution of 2 mg of
2
80 °C, capillary voltage at 37 V, tube lens offset at 50 V, and ion
spray voltage at 5 V.
homophymine A (1) was treated with 2.1 mg of NaOMe in dry
methanol (200 µL) at room temperature for 24 h. The reaction
mixture was neutralized with 0.1 N HCl, poured into ice-water,
and then extracted with n-BuOH. The n-BuOH phase was evapo-
rated under reduced pressure and the crude product (0.8 mg) was
subjected to ESI Q/TOF analysis. Opened methyl ester m/z 1630.74
Retention times (min) of FDAA-amino acids are given in
parentheses: D-Pip (26.3), L-Pip (27.8), D-Leu (36.5), L-Leu (31.3),
D-alloThrOMe (23.1), L-alloThrOMe (18.3), D-ThrOMe (24.8),
L-ThrOMe (19.6), D-Orn (8.6), and L-Orn (11.1).
To determine the absolute configuration of 3,4-diMeGln an
authentic sample of callipeltin A was used as standard. The
hydrolysate of callipeltin A contained (2S,3S,4R)-3,4-diMe-Glu
+
[M + H] .
Preparation of L-ThrOMe and L-alloThrOMe. A solution of
50 mg of N-Boc-L-Thr was treated with a large excess of
diazomethane in dry dichloromethane. The yellow solution was
stirred at room temperature for 30 min and then it was concentrated
under reduced pressure to give a residue (50 mg) that was subject
to the next reaction without purification. 2,6-Di-tert-butylpyridine
(1.4 mL) and methyl trifluoromethanesulfonate (0.7 mL) were added
sequentially to a solution of N-Boc-L-Thr methyl ester (0.2 mmol)
(
20.5).
The hydrolysate of homophymine A (1) contained L-Pip (28.3),
D-Orn (8.9), L-Leu (31.5), L-ThrOMe (19.6), and (2S,3S,4R)-3,4-
diMe-Glu (20.6).
To determine the absolute configuration of 2-amino-3-hydroxy-
,5-dimethylhexanoic acid (AHDMHA, 4), two aliquots of the
4
hydrolysate mixture were derivatized with L- and D-FDAA,
respectively, and then they were subjected to LC-MS as described
above. Retention times (min): L-FDAA-AHDMHA (33.5) and
D-FDAA-AHDMHA (28.1).
in CH
allowed to warm to room temperature where stirring was continued
for 14 h. Saturated NaHCO solution was added and the organic
phase was washed with water, dried (MgSO ), and then concentrated
2 2
Cl at 0 °C under argon atmosphere. The mixture was
3
Chiral HPLC Analysis. The acid hydrolysate of 1 (aliquot of
4
1
0 µL) was analyzed by chiral HPLC on a Phenomenex D-
in vacuo. Purification by column chromatography on silica gel with
n-hexane:EtOAc (99:1) as eluent gave the methyl ether in quantita-
tive yield as a colorless oil. A 1 mg portion of N-Boc-L-ThrOMe
methyl ester was dissolved in degassed 6 N HCl (0.5 mL) in an
evacuated glass tube and heated at 120 °C for 16 h. The solvent
penicillamine column [Chirex phase 3126 (D) (150 × 4.6 mm)].
The identity of serine in the acid hydrolysate was confirmed by
comparison of its retention times with those of authentic standards,
using HPLC under the following conditions: mobile phase, 2 mM
5
326 J. Org. Chem. Vol. 73, No. 14, 2008

 Zampella, Angela
Zampella, Angela