6
008
S. L. Capim et al. / Bioorg. Med. Chem. 21 (2013) 6003–6010
aromatic); 1735 (C@O of ester); 1681 (C@O of amide); 2924 (C–H
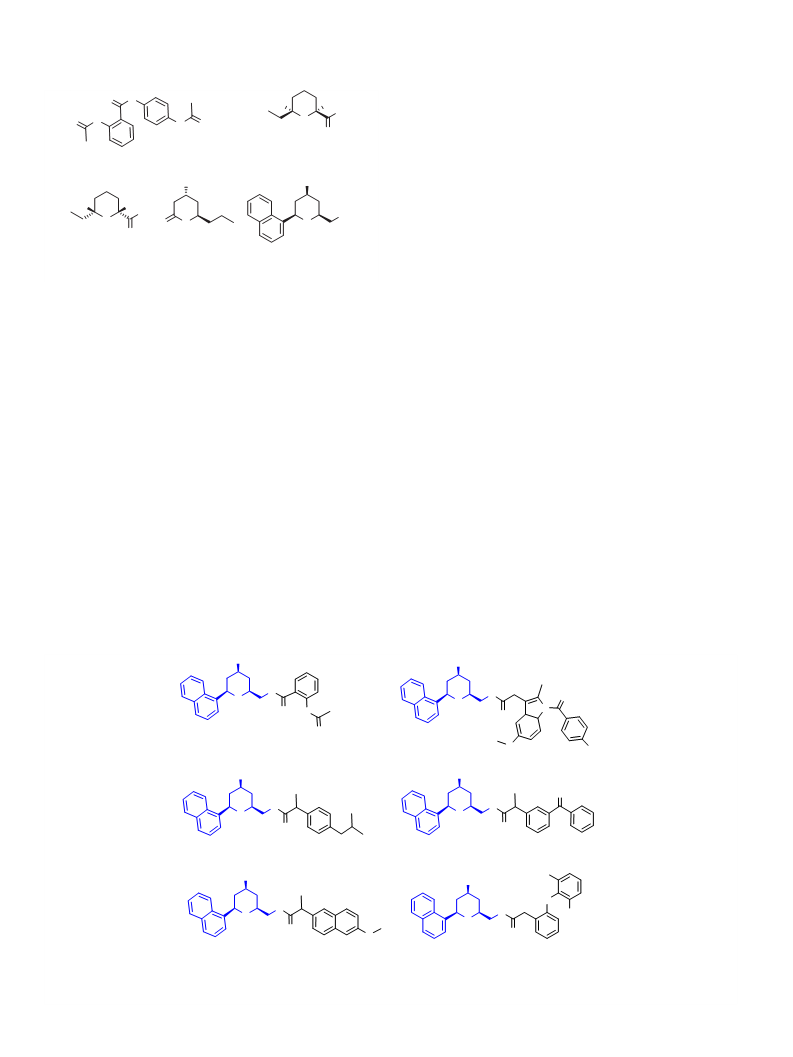
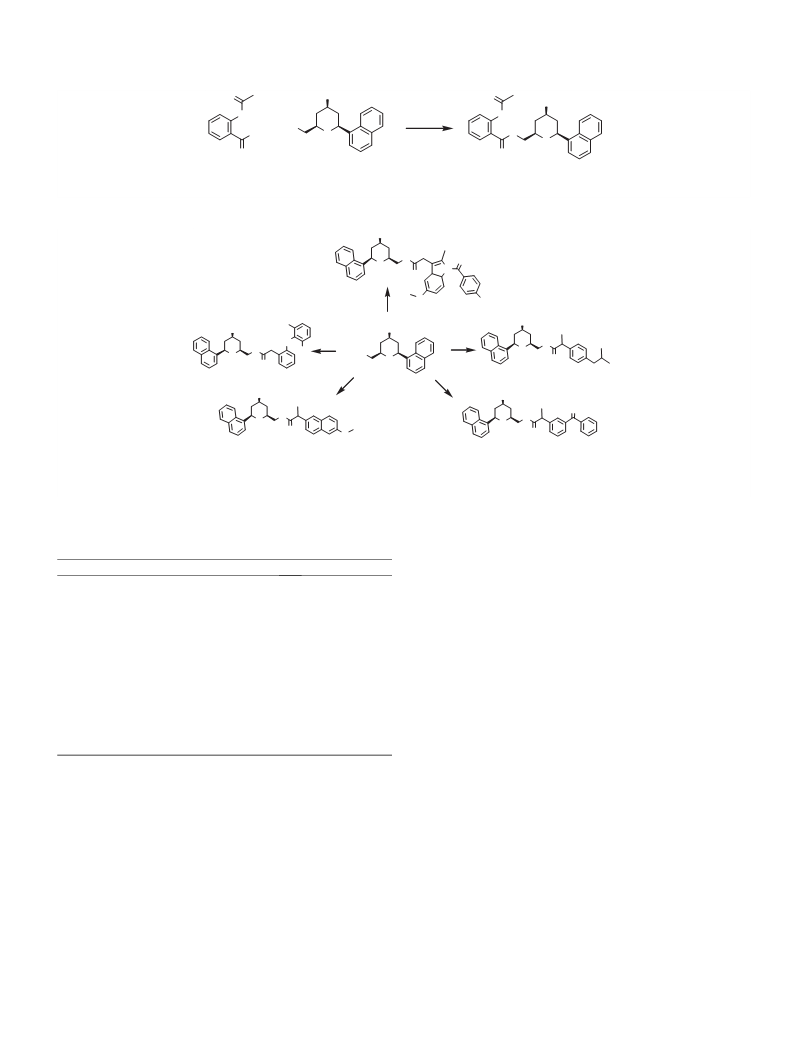
4.1.6. Synthesis of cis-(± ±-4-chloro-6-(naphthalen-1-yl±-
tetrahydro-2H-pyran-2-yl±methyl 2-(6-methoxynaphthalen-2-
yl±propanoate (8±
3
1
sp ); 3429 (N–H). H NMR (CDCl
sxt, 2H, J = 12 Hz); 2.12 (m, 2H); 2.36 (s, 3H); 2.55 (dd, 1H,
J = 4 Hz); 3.68 (t, 2H, J = 4 Hz); 3.72 (m, 3H); 3.86 (m, 1H); 4.06
3
, 400 MHz) d: 1.25 (t, 1H); 1.81
(
The reaction was performed by stirring a solution of naproxen
(
(
4
1
1
m, 1H); 4.24 (m, 2H, J = 4 Hz); 5.09 (m, 1H); 6.79 (m, 2H); 7.66
2
(2.0 g) in SOCl (5.0 mL) and the mixture was stirred at room temꢀ
1
3
m, 12H). C NMR (101 MHz; CDCl
3
) d: 13.37; 30.18; 38.24;
perature at 80 °C for 3 h. After the end of reaction (verified by chroꢀ
matography using hexane:ethyl acetate (7:3) as solvent), the
resulting mixture on evaporation afforded naproxen chloride in
95% yield. 1 mmol of naproxen chloride and 1 mmol of alcohol
(±)ꢀ3 were stirred in 10 mL of THF and 0.5 mL of TEA at 80 °C for
7 days. After the end of reaction (verified by chromatography),
2.77; 54.97; 55.52; 66.59; 74.92; 75.64; 101.23; 111.53;
12.31; 114.92; 122.74; 125.50; 128.94; 128.99; 130.49;
31.06; 133.64; 133.73; 136.01; 155.94; 168.17; 170.59. Anal.
Calcd for C35
6
H33Cl
2 5
NO , C, 68.18; H, 5.07; N, 2.27. Found: C,
8.19; H, 5.08; N, 2.22.
the resulting mixture was extracted with CH
2
Cl
2
(3 Â 20 mL). Pure
4
.1.4. Synthesis of cis-(± ±-4-chloro-6-(naphthalen-1-yl±-
product was obtained from the crude reaction by column chromaꢀ
tography through silica gel, using AcOEt:hexane as solvent in a raꢀ
tio of 3:7. The solvent mixture was concentrated under reduced
tetrahydro-2H-pyran-2-yl±methyl 2-(4-isobutylphenyl±
propanoate (6±
À1
The reaction was performed by stirring a solution of ibuprofen
pressure to afford 8 in 74% yield; IR (KBr, cm ): 825 and 800
(
9.7 mmol) in SOCl
2
(5.0 mL) and the mixture was stirred at room
(C–H aromatics); 1327–1178 (C–O); 1600 and 1500 (C@C aromatꢀ
ics); 1730 (C@O ester); 2970 (C–H sp ). H NMR (CDCl , 400 MHz)
3
d: 0.84 (t, 2H); 1.24 (m, 1H); 1.59 (m, 5H); 1.71 (m, 2H); 3.44 (m,
1H); 3.87 (m, 1H); 4.01 (s, 3H); 4.04 (m, 1H); 4.10 (m, 2H); 7.28
(m, 2H); 7.52 (m, 4H); 7.70 (m, 5H); 8.17 (d, 2H). 13C NMR
3
1
temperature at 80 °C for 3 h. After the end of reaction (verified by
chromatography), the resulting mixture evaporated obtained 95%
of ibuprofen chloride. 1 mmol of ibuprofen chloride and 1 mmol
of alcohol (±)ꢀ3 were stirred in 10 mL of THF and 0.5 mL of TEA
at 80 °C for 7 days. After the end of reaction (verified by chromaꢀ
tography using hexane:ethyl acetate (7:3) as solvent), the resultꢀ
3
(101 MHz; CDCl ) d: 10.29; 18.32; 18.43; 21.91; 25.93; 28.97;
44.41; 45.30; 56.97; 63.98; 66.44; 113.88; 113.94; 116.73;
123.88; 123.97; 126.12; 126.13; 127.39; 127.49; 127.81; 129.45;
129.50; 130.99; 131.03; 136.41; 136.63; 152.75; 174.50. Anal.
ing mixture was extracted with CH
2
Cl
2
(3 Â 20 mL). Pure product
was obtained from the crude reaction by column chromatography
through silica gel, using AcOEt:hexane as solvent at a ratio of 3:7.
The solvent mixture was concentrated under reduced pressure to
afford 6 in 70% yield; IR (KBr, cm ): 779 and 736 (C–H aroꢀ
matic); 1377–1330 (C–O); 1512 and 1454 (C@C aromatic); 1732
4
Calcd for C32H29ClO : C, 74.92; H, 5.70. Found: C, 74.85; H, 5.62.
À1
4.1.7. 4.1.7.Synthesis of cis-(± ±-4-chloro-6-(naphthalen-1-yl±-
tetrahydro-2H-pyran-2-yl±methyl 2-(2-(2,6-dichlorophenylamino±
phenyl±acetate (9±
3
1
(
C@O of ester); 2954 (C–H sp ). H NMR (CDCl
3
, 400 MHz) d:
0
2
1
3
3
1
1
.86 (dd, 6H, J = 4 Hz); 1.51 (dd, 3H); 1.92 (m, 5H); 2.38 (dd,
H, J = 4 Hz); 2.52 (m, 1H); 3.81 (m, 2H); 4.26 (m, 3H); 5.05 (d,
The reaction was performed by stirring a solution of alcohol 3
(0.53 mmol) in CH Cl (2.0 mL), triethylamine (0.2 mL), 4ꢀtolueneꢀ
2 2
sulfonyl chloride (1.3 mmol) and the mixture was stirred at 0 °C for
30 min. and 12 h at room temperature. After the end of reaction
(verified by chromatography using hexane:ethyl acetate (7:3) as
H, J = 12 Hz); 7.01 (m, 2H); 7.22 (dd, 2H, J = 4 Hz); 7.53 (m,
H); 7.89 (m, 3H). 1 C NMR (101 MHz; CDCl
3
3
) d: 18.31; 22.40;
0.11; 38.32; 42.87; 55.11; 66.18; 66.33; 75.04; 75.70; 122.88;
23.33; 125.54; 126.14; 127.18; 128.40; 128.96; 129.29;
30.13; 133.72; 136.17; 136.24; 137.42; 140.60, 174.51. Anal.
2 2
solvent), the resulting mixture was extracted with CH Cl
(3 Â 20 mL). The organic phase was dried with anhydrous sodium
sulfate and concentrated under reduced pressure where tosylate
derivative of alcohol 3 was obtained a 95% yield. After, 0.1 mmol
of tosyl derivative of alcohol 3 was stirred with 0.15 mmol of dicꢀ
Calcd for C29
3
H33ClO , C, 74.90; H, 7.62. Found: C, 74.86; H, 7.57.
4
.1.5. Synthesis of cis-(± ±-4-chloro-6-(naphthalen-1-yl±-
tetrahydro-2H-pyran-2-yl±methyl 2-(3-benzoylphenyl±
propanoate (7±
The reaction was performed by stirring a solution of ketoproꢀ
2
fen (2.0 g) in SOCl (5.0 mL) and the mixture was stirred at room
temperature at 80 °C for 3 h. After the end of reaction (verified by
chromatography), the resulting mixture on evaporation afforded
ketoprofen chloride in 95% yield. 1 mmol of ketoprofen chloride
and 1 mmol of alcohol (±)ꢀ3 were stirred in 10 mL of THF and
2 3
lofenac Potassium, 22 mg of K CO in 1 mL of DMF under microꢀ
wave irradiation (100 W) at 100 °C for 90 min. Pure product was
obtained from the crude reaction by column chromatography
through silica gel, using AcOEt:hexane as solvent in a ratio of
3:7. The solvent mixture was concentrated under reduced pressure
À1
to afford 9 in 90%; IR (KBr, cm ): 777 and 742 (C–H aromatics);
1303 (C–O); 1577 and 1502 (C@C aromatics); 1691 (C@O ester);
3
1
3
2918 (C–H sp ); 3348 (N–H). H NMR (CDCl , 400 MHz) d: 1.26
0
.5 mL of TEA at 80 °C for 7 days. After the end of reaction (veriꢀ
fied by chromatography using hexane:ethyl acetate (7:3) as solꢀ
vent), the resulting mixture was extracted with CH Cl
(t, 1H, J = 8 Hz); 1.81 (dd, 1H, J = 12 Hz); 2.09 (q, 2H, J = 12 Hz);
2.23 (m, 1H); 2.58 (t, 1H, J = 4 Hz); 2.61 (t, 4H, J = 4 Hz); 3.76 (m,
4H); 4.33 (m, 1H); 5.14 (d, 1H); 7.23 (m, 2H); 7.51 (m, 3H); 7.62
(d, 1H, J = 8 Hz); 7.85 (m, 2H); 8.00 (d, 1H, J = 8 Hz). 13C NMR
(101 MHz; CDCl ) d: 38.02; 38.15; 42.76; 55.39; 65.66; 75.70;
3
77.91; 118.24; 122.04; 122.89; 123.26; 123.90; 123.98; 125.37;
125.64; 126.29; 128.05; 128.60; 128.82; 128.96; 129.38; 130.32;
2
2
(
3 Â 20 mL). Pure product was obtained from the crude reaction
by column chromatography through silica gel, using AcOEt:hexꢀ
ane as solvent at a ratio of 3:7. The solvent mixture was concenꢀ
trated under reduced pressure to afford 7 in 72% yield; IR (KBr,
À1
cm ): 786 and 721 (C–H aromatics); 1315–1172 (C–O); 1593
130.90; 133.73; 136.09; 142.60; 175.93. Anal. Calcd for C30
H26Cl3ꢀ
and 1450 (C@C aromatics); 1658 (C@O) 1735 (C@O ester); 2958
NO : C, 64.94; H, 4.72; N, 2.52. Found: C, 65.07; H, 4.71; N, 2.51.
3
3
1
(
2
(
1
1
7
1
1
C–H sp ). H NMR (CDCl
.48 (m, 1H); 2.73 (m, 3H); 2.91 (m, 2H); 3.11 (m, 1H); 3.39
m, 1H); 3.74 (m, 1H); 4.66 (m, 1H); 5.01 (m, 2H); 5.31 (m,
3
, 400 MHz) d: 2.10 (d, 1H, J = 6 Hz);
4.2. Pharmacology
4.2.1. Animals
The experiments were carried out on male Albino–Swiss mice
(body weight 20–24 g). The animals were housed in wire mesh
cages in a room temperature and exposed to a 12 h light:12 h dark
cycle. The animals had free access to standard pellet diet, tap water
was given ad libitum. The protocol for this study was approved by
H); 5.47 (m, 2H); 8.73 (m, 16H). 13C NMR (101 MHz; CDCl
3
) d:
9.74; 27.37; 30.37; 39.75; 44.24; 46.67; 56.41; 65.48; 67.88;
6.32; 124.23; 124.60; 126.93; 127.52; 129.66; 129.81; 130.33;
30.52; 131.43; 132.93; 133.86; 135.08; 137.49; 138.81;
39.19; 142.03; 142.20; 175.27; 197.82. Anal. Calcd for C32
: C, 74.92; H, 5.70. Found: C, 74.85; H, 5.62.
H
29ꢀ
ClO
4

 Capim, Saulo L.
Capim, Saulo L.