phile combinations.4 Several other groups have described
similar methodologies with either silyl- or boryl-derived
electrophiles.5 It appears, however, that in situ trapping has
not been widely applied to arene halogenation.3d,e Further-
more, potassium phosphate and tBuOLi have not been
employed as metalating agents for such reactions. We report
here a simple method for in situ halogenation of sp2 C-H
bonds in heterocycles and electron-deficient arenes under
noncryogenic conditions.
Table 1. Halogenation Scopea
Minor modifications of the copper-catalyzed arylation
conditions allowed the development of an efficient process
for in situ sp2 C-H bond halogenation. The scope of the
halogenation reaction is presented in Table 1. Electron-rich
heterocycles such as benzothiazole, butylimidazole, and
phenyloxazole can be chlorinated and brominated (entries
1-4). The halogenating reagent must be optimized for each
case. Either carbon tetrahalides (entries 1-3) or dibromo-
tetrafluoroethylene (entry 4) may be employed for electron-
rich heterocycle halogenation.
Electron-deficient heterocycles such as pyridine N-oxide
and 2-phenylpyridine N-oxide are brominated by employing
a mixture of tBuOLi base and carbon tetrabromide (entries
5 and 6). For pyridine N-oxide, dihalogenation/reduction to
the dibromopyridine product is obtained in a moderate yield
(entry 6). Selective monohalogenation of pyridine N-oxide
was not achieved under a variety of conditions. In contrast
with the copper-catalyzed arylation process, halogenation is
unsuccessful for pyridazine and pyrimidine, presumably due
to their insufficient acidity.6 Consequently, higher acidity is
required for the halogenation reaction if it is compared to
arylation.7
Electron-deficient arenes can also be efficiently halogen-
ated (entries 7-14). Pentachlorobenzene is iodinated in
excellent yield by using a combination of iodine(I) chloride
and tBuOLi in DMF (entry 7). Nitro and cyano groups are
tolerated as shown in bromination of 3-nitrofluorobenzene
(entry 8) and one-step tribromination of 3,5-difluorobenzo-
nitrile (entry 9). For the latter substrate, selective mono-
halogenation could not be achieved. If acidic pentafluoro-
benzene is halogenated, even the weak K3PO4 base can be
employed (entry 10), affording iodopentafluorobenzene in
excellent yield. Previously, butyllithium has been used for
pentafluorobenzene deprotonation-reaction with electrophiles
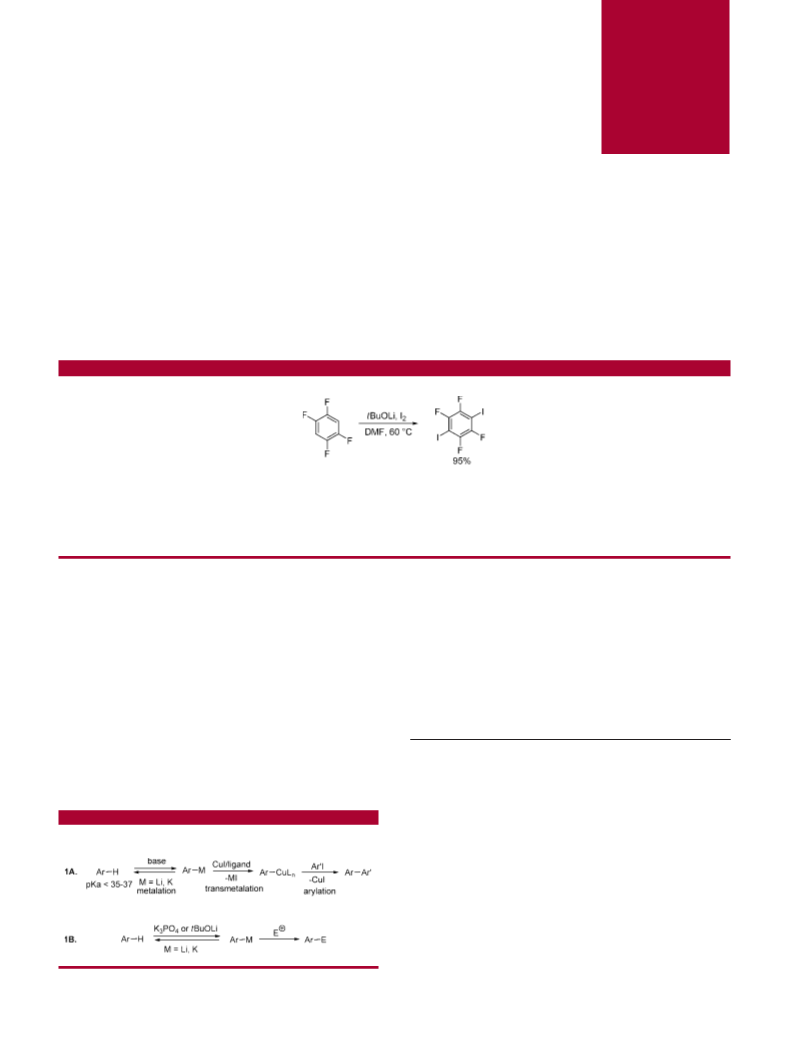
sequences.8 Monoiodination of methoxytetrafluorobenzene
and one-step diiodination of 1,2,4,5-tetrafluorobenzene af-
(4) (a) Dua, S. S.; Gilman, H. J. Organomet. Chem. 1974, 64, C1-C2.
(b) Howells, R. D.; Gilman, H. Tetrahedron Lett. 1974, 15, 1319. (c)
Schlosser, M.; Guio, L.; Leroux, F. J. Am. Chem. Soc. 2001, 123, 3822.
(d) Imahori, T.; Kondo, Y. J. Am. Chem. Soc. 2003, 125, 8082. (e) Eaton,
P. E.; Cunkle, G. T.; Marchioro, G.; Martin, R. M. J. Am. Chem. Soc. 1987,
109, 948.
(5) (a) Vazquez, E.; Davies, I. W.; Payack, J. F. J. Org. Chem. 2002,
67, 7551. (b) Caron, S.; Hawkins, J. M. J. Org. Chem. 1998, 63, 2054. (c)
Kristensen, J.; Lyse´n, M.; Vedsø, P.; Begtrup, M. Org. Lett. 2001, 3, 1435.
(d) Black, W. C.; Guay, B.; Scheuermeyer, F. J. Org. Chem. 1997, 62,
758.
a Substrate (1 equiv), base (2-4 equiv), halogenating reagent (1.5-4
equiv). Yields are isolated yields. See Supporting Information for details.
b m-Xylene solvent. c 1,3,5-Trifluorobenzene (3 equiv), ICl (1 equiv).
b 1,3,5-Trifluorobenzene (1 equiv), ICl (4 equiv).
(6) (a) Shen, K.; Fu, Y.; Li, J.-N.; Liu, L.; Guo, Q.-X. Tetrahedron 2007,
63, 1568. (b) Bordwell, F. G. Acc. Chem. Res. 1988, 21, 456.
(7) A figure listing substrates not halogenated under these conditions is
included in Supporting Information.
fords the corresponding halides in excellent yields (entries
11 and 12). 1,3,5-Trifluorobenzene can be either mono-
(8) Review Burton, D. J.; Yang, Z.-Y.; Morken, P. A. Tetrahedron 1994,
50, 2993.
422
Org. Lett., Vol. 11, No. 2, 2009

 Do, Hien-Quang
Do, Hien-Quang