Inter-Ring Torsions in N-Phenylmaleimide
J. Phys. Chem. A, Vol. 103, No. 32, 1999 6411
TABLE 4: Inter-ring Torsion Angles (Degrees) from
Semiempirical and ab Initio [Level: 6-31G(d,p)] Minimum
Energy Conformers Compared to the Experimental
Crystallographic Values
A comparison between the solution (Figure 1) and calculated
spectra (Table 3) provides some qualitative agreement. Ignoring
for the moment the o-iodo derivative, two important features
are seen in the spectra above 250 nm for N-phenylmaleimide
and its three lighter o-halophenyl derivatives. Calculations
predict two (for Cl and Br derivatives) or three (for the parent
and F derivative) transitions in the same range. The oscillator
strengths of the intermediate band in the parent and fluoro
derivative are very small, leaving essentially two important
features in each case. The lower energy bands are calculated to
shift slightly to shorter wavelength in the halogen-substituted
systems in the order H < F < Cl ≈ Br, and the solution spectra
of the series follow this trend. The next higher of the two
observed ultraviolet bands in the solution spectra are found at
260-270 nm. Calculations suggest that these next highest
intensity bands have π f π* (phenyl) character and should
remain at 270 nm for H through Br and that the oscillator
strength for the fluoro derivative should be larger than the others.
Both the position and intensities of the observed bands are
roughly in agreement with the calculations.
theorya
compound
X-ray
(o-substituted) AM1 SCF DFT-B3LYP MP2 experiment
H
F
Cl
Br
I
26
49
79
85
90
52.7
69.5
87.6
39.8
56.2
71.2
44.3 49.5
56.4 54.2, 66.8b
67.6 66.1
71.1
83.9
a Vacancies not modeled due to basis sets limitations. b Two
conformers in the crystal.
in N-(2′-iodophenyl)maleimide which might represent overlap
of bands. The intensity of the longer wavelength band seems
too strong to be purely an n f π* transition, and that of the
shorter wavelength band seems to be too weak to be purely a
π f π* transition.
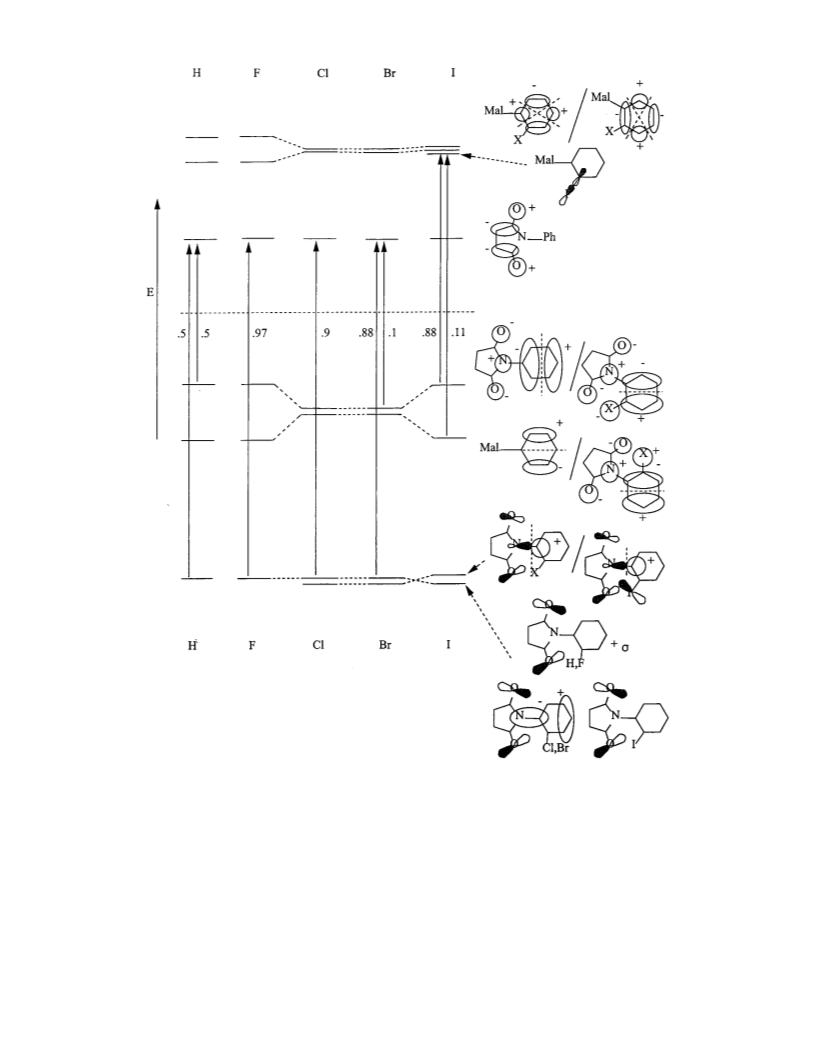
Gas-phase electronic spectra for the series have been calcu-
lated by the Zindo/S CIS method at the AM1 minimum energy
conformers. The results associated with the lowest energy
transition is illustrated in Figure 4 which depicts the six or seven
frontier molecular orbitals for each compound in the series. The
lowest unoccupied molecular orbital in all members of the series
is localized on the maleimide ring and may be described as
having π*(CdC) and π*(CdO) character. Above this in energy,
the next two are π* orbitals localized on the phenyl rings and
each having two local perpendicular nodal planes. These two
differ somewhat in energy in the parent and o-fluoro compounds,
but have nearly the same energy in the rest of the series. In the
o-iodo derivative, a σ*(C-I, 2p-5p) orbital becomes nearly
equal in energy to the π*(phenyl) orbitals. In the occupied
regime, the two highest occupied molecular orbitals have
n (O,N,X) + π(phenyl) character, each with one local perpen-
dicular phenyl nodal plane. These two differ somewhat in energy
in the parent, o-fluoro, and o-iodo compounds, but have nearly
the same energy in the o-chloro and o-bromo derivatives. Below
this, each compound has a n (O,N) orbital which additionally
has significant σ(C-C, C-N, C-H) character for the parent
and the o-fluoro compounds, changing to π(C-N) and π(phen-
yl) character for the o-chloro and o-bromo derivatives and
modifying further to π(C-N) and π(phenyl) character in the
o-iodo derivative. These are depicted in Figure 4.
Beginning with AM1 molecular geometries, the electronic
spectra were calculated for excited singlets with Zindo/S
configuration interaction limited to single excitation (CIS) out
of the Hartree-Fock ground states for each member of the
series. The long-wavelength band assignments are given in
Figure 4, and for the parent and three lightest o-halo derivatives
involves a transition from an orbital with n (O,N) character to
the π*(maleimide) orbital (LUMO). The transition originates
from a mixture of orbitals having n (O,N) and π(phenyl)
character (HOMO) in the parent and o-bromo cases. The lower
energy orbital in this transition decreases in σ and increases in
π character as the inter-ring angle increases. In the o-chloro
and o-bromo cases, a second orbital with n (O,N) and π*(C-
N) character also contributes. The nonbonded character of the
longer wavelength transitions is supported by the hypsochromic
shift in this band observed in the spectra of the o-fluoro
derivative upon progression to more polar solvents. A change
is predicted for the lower energy bands of the o-iodo derivative.
The long-wavelength transition appears from higher occupied
molecular orbitals with n (O,N) and π(phenyl) character to an
unoccupied one with σ(C-I, 2p-5p) character.
For the o-iodo derivative, a change is seen in the observed
spectrum (Figure 1) in which the stronger, shorter wavelength
band appears to shift slightly to longer wavelength compared
to the lighter halogen derivatives, and to intensify. Computations
suggest that this may be accounted for by a change in the
transition responsible for the longer wavelength absorption
compared to the parent and lighter halogen derivatives. The
longer wavelength band seems to be due to a πPh f σ*(C-I,
2p-5p) transition and to be accompanied by an increase in
intensity and shift to slightly longer wavelength compared to
the other o-halo derivatives. These predictions are in accord
with the spectrum of the N-(o-iodophenyl)maleimide.
4. Conclusions
The structures of N-phenylmaleimide and the o-halo-N-
phenylmaleimides (fluoro, chloro, bromo, and iodo) have been
determined and show that the inter-ring torsion increases with
increasing steric size of the halogen. Semiempirical AM1 and
several ab initio calculations generally agree with the solid-
state structures. The AM1 computations show a broad and
shallow deformation energy associated with the inter-ring
twisting for all members of the series. This deformation leads
to a modest barrier to rotation at the orthogonal inter-ring
conformation for the parent N-phenylmaleimide and shifts to
conformations with lower twist angles for any of the N-(o-
halophenyl)maleimides. Considerable maleimide and phenyl
distortions are expected for the chloro, bromo, and iodo
derivatives as the inter-ring torsion angle is reduced; for these
molecules and for the fluoro derivative, torsional interconversion
proceeds through the perpendicular conformation. Calculations
also support assignment of the ultraviolet absorption features
above 250 nm to several phenyl and maleimide ring transitions.
Except for the o-iodo derivative, the shorter wavelength features
appears to be principally a phenyl π f π* transition, while the
longer wavelength feature has {n (O,N) + πPh} f π*Mal
character. Longer wavelength absorptions in the o-iodo deriva-
tive may arise from overlapping n (O,N) + πPh f σ(C-I, 2p-
5p) transitions.
Acknowledgment. We thank Mr. C. Jace Pugh for experi-
mental assistance, Dr. Jeffrey D. Zubkowski, Jackson State
University, for access to diffraction equipment, and Dr. William
A. Parkinson, Southeastern Louisiana University, for assistance
with computations. We acknowledge support from First Chemi-

 Miller, Christopher W.
Miller, Christopher W.