4
A.J. Roche / Journal of Fluorine Chemistry 175 (2015) 1–5
spectra were performed at ambient temperature in deuterated
acetone at 282 MHz and 300 MHz, respectively, except where
indicated in the text. Chemical shifts for 19F and 1H spectra were
determined relative to CFCl3 (0.0 ppm) and TMS (0.0 ppm),
respectively. All products were colourless solids, except where
specified otherwise. All reagents, unless otherwise specified, were
used as purchased from Aldrich or Fisher. Column chromatography
was performed using chromatographic silica gel 200–425 mesh, as
supplied by Fisher. Low-resolution mass spectrometry was
performed at the Center for Advanced Food Technology, New
Brunswick, NJ, the University of Pennsylvania, Philadelphia, PA,
and at Rutgers University-Camden. High-resolution mass spec-
trometry was performed at the University of Pennsylvania,
Philadelphia, PA. The starting materials (OFP-I 2, OFP-Br 4, OFP-
Cl 3 and PCP-Br) were prepared according to their published
literature procedures [13,41].
etherate (3.30 g, 12.80 mmol) was stirred at room temperature
under a nitrogen atmosphere for 1 h. Then a hexane solution of
tert-butyl lithium (1.7 M, 6.6 ml, 11.22 mmol) was added
dropwise, and the mixture was stirred for 2 h at room
temperature. The resulting pale brown solution was then used
in the following reaction: a degassed THF solution (20 ml)
containing
phane
4-iodo-1,1,2,2,9,9,10,10-octafluoro[2.2]paracyclo-
(1.53 g, 3.320 mmol) and palladium dichloride
2
(110 mg, 0.62 mmol) was stirred at room temperature under a
nitrogen atmosphere for 1 h. The pre-formed PCP-MgBr solution
(27 ml, 11.22 mmol) was slowly added dropwise so that
complete addition took 2 h. The resulting black solution was
stirred for another 6 h. After that time, evaporation of the
solvent was followed by the addition of ice water (200 ml), and
the precipitated solids were chromatographed on silica gel
(hexane/dichloromethane 8.5/1.5) to give (Rf = 0.49) octafluor-
o[2.2]paracyclophane
1
(701 mg, 60%), and (Rf = 0.30) 4,40-
4.1. Synthesis of 4-phenyl-1,1,2,2,9,9,10,10-
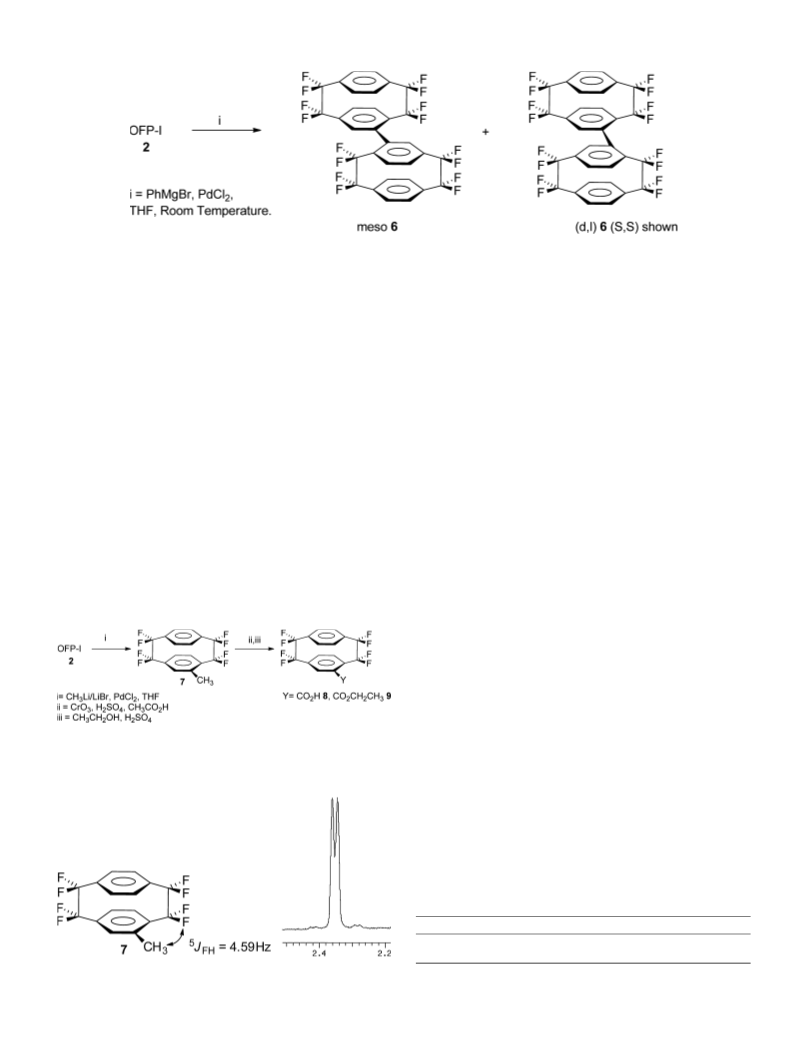
bis(1,1,2,2,9,9,10,10-octafluoro[2.2]paracyclophane) 6 (350 mg,
30%). The ratio of diastereomers of 6 was 1.5:1 (meso:dl) as
established by 19F NMR, and GC analysis.
octafluoro[2.2]paracyclophane 5
A
degassed THF solution (4 ml) containing 4-iodo-
1,1,2,2,9,9,10,10-octafluoro[2.2]paracyclophane (640 mg,
2
4.3. Synthesis of 4-methyl-1,1,2,2,9,9,10,10-
1.34 mmol) and palladium dichloride (8 mg, 0.04 mmol) was
stirred and brought to reflux under a nitrogen atmosphere. A THF
solution of phenyl magnesium bromide (1 M, 4 ml, 4.0 mmol)
was added via syringe and syringe pump, with completion taking
4 h, and the black solution was refluxed overnight. Evaporation
of the solvent was followed by the addition of ice water (75 ml),
and the precipitated solids were chromatographed on silica gel
(hexane/dichloromethane 8.5/1.5) to give (Rf = 0.49) octafluor-
o[2.2]paracyclophane 1 (38 mg, 8%) and (Rf = 0.39) 4-phenyl-
octafluoro[2.2]paracyclophane 7
A
degassed THF solution (100 ml) containing 4-iodo-
1,1,2,2,9,9,10,10-octafluoro[2.2]paracyclophane (10.5 g,
2
21.87 mmol) and palladium dichloride (0.216 g, 1.22 mmol)
was stirredunderanatmosphere ofnitrogen, and cooled to0 8C inan
ice bath. Over a period of 2 h, an ether solution of methyl lithium/
lithium bromide complex (1.5 M, 44 ml, 66.9 mmol) was added
dropwise. The solution was maintained at 0 8C for 3 h, and then
allowed to warm to room temperature. Analysis of the crude
mixture by 19F NMR against an internal standard of trifluorotoluene
showed the mixture to comprise of 4-methyl-1,1,2,2,9,9,10,10-
Octafluoro[2.2]paracyclophane 7 (17.71 mmol, 81%) and octafluor-
oparacyclophane 1 (3.72 mmol, 17%). Moist acetone (100 ml) was
carefully added to the reaction mixture, and this mixture was
filtered, and evaporated under reduced pressure. The resulting solid
was suitable for further reactions.
1,1,2,2,9,9,10,10-octafluoro[2.2]paracyclophane
5
(476 mg,
83%). Products 1 and 5 were identified by comparison of their
19F and 1H NMR spectra, and GCMS analyses, with published
literature data [8,13], and additionally were found to be identical
to authentic samples previously prepared. See Supporting
information for spectra and data.
4.2. Synthesis of 4,40-bis(1,1,2,2,9,9,10,10-
octafluoro[2.2]paracyclophane) 6
An analytically pure sample was obtained by chromatogra-
phy on silica gel (hexane/dichloromethane 19/1) to give
(Rf = 0.45) 4-methyl-1,1,2,2,9,9,10,10-octafluoro[2.2]paracyclo-
phane 7; mp = 126–131 8C. (NMR spectra obtained in CDCl3 due
4.2.1. Method A
A
degassed THF solution (10 ml) containing 4-iodo-
1,1,2,2,9,9,10,10-octafluoro[2.2]paracyclophane 2(1.60 g, 3.35 mmol)
and palladium dichloride (20 mg, 0.10 mmol) was stirred at room
temperature under a nitrogen atmosphere for 1 h. A THF solution of
phenyl magnesium bromide (1 M, 10 ml, 10.0 mmol) was added via
syringe and syringe pump, so that complete addition took 5 h. The
resulting black solution was stirred for another 6 h. After that time,
evaporation of the solvent was followed by the addition of ice
water (250 ml), and the precipitated solids were chromato-
graphed on silica gel (hexane/dichloromethane 8.5/1.5) to give
to superior solubility of OFP-CH3). 1H NMR 7.39 (d, 3J = 8.70 Hz,
d
1H); 7.41 (d, 3J = 8.70 Hz, 1H); 7.26–7.04 (m, 4H); 6.98 (d,
3J = 8.70 Hz, 1H); 6.75 (s, 1H); 2.35 (d, 5J = 4.59 Hz, 3H); 19F NMR
d
ꢂ110.21 (dm, 2J = 243.3 Hz, 1F); ꢂ111.97 (d, 2J = 245.3 Hz, 1F);
ꢂ113.02 (d, 2J = 239.1 Hz, 1F); ꢂ113.61 (d, 2J = 239.1 Hz, 1F);
ꢂ116.41 (d, 2J = 239.1 Hz, 1F); ꢂ116.91 (d, 2J = 237.1 Hz, 1F);
ꢂ116.52 (d, 2J = 237.1 Hz, 1F); ꢂ117.03 (d, 2J = 239.1 Hz, 1F); MS
m/z 366 (M+, 20%), 190 (100), 176 (21). Anal. Calcd for C17H10F8:
C, 55.70.46; H, 2.73. Found: C, 55.42; H, 2.55.
(Rf = 0.49) octafluoro[2.2]paracyclophane
1
(177 mg, 15%),
(Rf = 0.39) 4-phenyl-1,1,2,2,9,9,10,10-octafluoro[2.2]paracyclo-
phane 5 (545 mg, 38%), and (Rf = 0.30) 4,40-bis(1,1,2,2,9,9,10,10-
octafluoro[2.2]paracyclophane) 6 (517 mg, 44%). The ratio of
diastereomers of 6 was 29:15 (meso:dl) as established by 19F NMR
and GC analysis using a J&W DB-5ms column (Retention times of
24.0 and 26.5 min. for dl and meso, respectively). Products 6 were
identified by comparison of their 19F and 1H NMR spectra, and
GCMS analyses, with published literature data [15]. See Support-
ing information for spectra and data.
4.4. Synthesis of 4-carboxy-1,1,2,2,9,9,10,10-
octafluoro[2.2]paracyclophane 8
Sulphuricacid (12 ml, 98%conc.) wascarefullyaddedtoasolution
of glacial acetic acid (400 ml) containing 4-methyl-1,1,2,2,9,9,10,10-
Octafluoro[2.2]paracyclophane 7 (6.64 g, 18.14 mmol). This mixture
was immersed in a cool water bath, and with vigorous stirring,
chromium trioxide (40.00 g, 44 mmol) was added over a period of
2 h, and left tostir for a further 2 h. Then the mixture was poured into
ice water (1000 ml), and left to stir overnight. The resulting solids
werefiltered, and analysisby 19F NMR against an internal standardof
trifluorotoluene showed the mixture to comprise of 4-carboxyl-
4.2.2. Method B
A degassed THF solution (20 ml) containing 4-bromo[2.2]par-
acyclophane (3.67 g, 12.80 mmol) and magnesium bromide
1,1,2,2,9,9,10,10-octafluoro[2.2]paracyclophane
8
(15.42 mmol,

 Roche, Alex J.
Roche, Alex J.