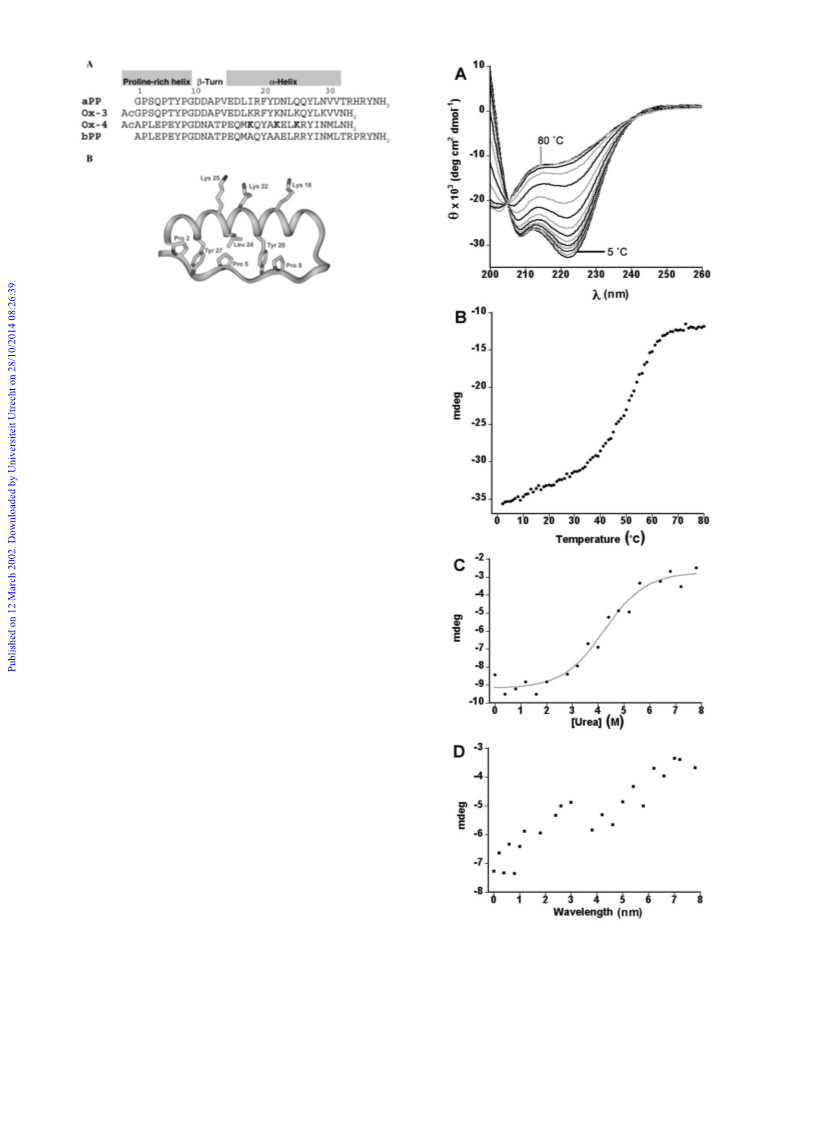
the unfolding reaction.14 The gradual decrease of the helical
content of Oxaldie-3 with increasing temperature was typically
that of a molten globule state. It is interesting to note that the
CD-spectra of both Oxaldie-3 and -4 are not those of fully
denatured proteins even at 80 ЊC (Fig. 2A).14
Similarly, Oxaldie-3 and Oxaldie-4 behaved qualitatively dif-
ferently in urea denaturation experiments. The denaturation
curve of Oxaldie-4 was sigmoidal as expected for a stably folded
protein (Fig. 2C). The concentration of urea at which 50% of
Oxaldie-4 was denatured was determined to be 4.26 M from the
denaturation curve. Experimentally it has been shown that
there is a linear relationship between the free energy of unfold-
ing a protein in the presence of urea and the concentration of
the denaturant,20–22 eqn. (1).
aliphatic residues of the α-helix confirmed the presence of a
tertiary structure similar to that of bPP (Fig. 1). The CD and
NMR data presented herein clearly showed that the conform-
ational properties of Oxaldie-3 and Oxaldie-4 in solution were
different. Oxaldie-4 exhibited stable secondary and tertiary
structure, resembling that of native bPP. Oxaldie-3, on the other
hand, was only loosely compacted and appeared to be in a
molten globule-like state.14 It is possible that in solution the
parent proteins avian and bovine PP also display similar differ-
ences in their conformational stability.
Catalytic properties of Oxaldie-4
The Oxaldie-4 catalysed decarboxylation of oxaloacetate was
studied by UV spectroscopy in a coupled enzymatic assay.1
Rates for the decarboxylation of oxaloacetate to produce pyru-
vate were obtained from measuring the rate of conversion of
NADH to NADϩ in the lactate dehydrogenase-mediated con-
version of pyruvate to lactate. The Oxaldie-4 catalysed reaction
observed Michaelis-Menten kinetics with kcat = 0.229 sϪ1 and
KM = 64.8 mM and a catalytic efficiency kcat/KM of 2.9 MϪ1 sϪ1
(Table 1). The catalytic efficiency of Oxaldie-4 was independent
of the peptide concentration for the whole range studied (4–40
µM) as would be expected for a peptide with enzyme-like
behaviour. The catalytic efficiency of Oxaldie-4 was improved
approximately twofold relative to Oxaldie-3 (Table 1). The fluc-
tuating tertiary structure of molten globules may have sug-
gested that improving the stability of the protein fold should
have led to a significant increase in the catalytic efficiency of the
peptide. However, because the molten globule state of Oxaldie-
3 is characterised by a compactness similar to that of the native
state with native-like secondary structure, the high flexibility of
the lysine side chains, which make up the active sites, is most
likely the explanation for the similar catalytic properties of
Oxaldie-3 and -4.
In summary, changing the backbone from that of avian to
that of bovine PP resulted in the conversion of a molten globule
(Oxaldie-3) to a stably folded peptide (Oxaldie-4) with a well-
defined three-dimensional structure with slightly improved
catalytic efficiency. It is commonly assumed that homologous
proteins adopt similar conformations in solution and the differ-
ent properties of Oxaldie-3 and -4 may therefore be surprising.
14 of the 31 amino acids are identical and several of the non-
identical residues are replaced in a conservative fashion. In
medium to large size proteins this level of similarity would be
considered quite high. However, due to the much smaller num-
ber of interactions that determine the folded structure of small
peptides such as the 31 residue Oxaldie-3 and -4, small changes
in their primary sequence can clearly have dramatic effects on
the stability of the folded conformation. Investigations are
under way to define the key determinants of the different sta-
bilities of Oxaldie-4 and Oxaldie-3 and indeed of bPP and aPP.
D
∆GU–F = ∆GUH–OF Ϫ mU–F[D]
(1)
2
From the measurement of the ellipticity at 222 nm for
Oxaldie-4 as a function of the concentration of urea, the free
H2O
U–F
energy of unfolding in water ∆G and mU–F, a constant which
is proportional to the increase in the degree of exposure of the
protein on denaturation, could be estimated as Ϫ2.76 kcal
molϪ1 and 0.65 kcal molϪ1, respectively. The behaviour of
Oxaldie-3 was very different and the free energy of unfolding
could not be determined.14 Its denaturation curve was not sig-
moidal. The ellipticity at 222 nm (and therefore the decrease of
the α-helical content) deceased in an almost linear fashion with
increasing concentrations of urea (Fig. 2D).
NMR experiments with Oxaldie-4 confirmed that the peptide
adopted a stably folded conformation. 1H Homonuclear
TOCSY and NOESY spectra of Oxaldie-4 in H2O were run
for resonance assignment and structural characterisation. The
complete sequence-specific assignment was assisted by use of
both the sequential amide proton NOEs and the close similarity
to the chemical shift assignment in bPP.12 In contrast with the
1H-chemical shifts observed for Oxaldie-3,14 a significant dis-
persion in the amide proton chemical shifts of Oxaldie-4, simi-
lar to the extent of dispersion in native bPP, was immediately
evident in the TOCSY experiment indicating the formation of
secondary structure. Most spin systems in Oxaldie-4 for amino
acids of a common type were resolved. Sequence-specific
assignment of the three lysine side chains, which were not pres-
ent in native bPP, was accomplished by NH–NH NOE correl-
1
ations. A 2D H,13C HSQC–TOCSY spectrum was useful for
confirming assignments, particularly using the characteristic
threonine 13C chemical shifts. Pro 8 in Oxaldie-4 exhibited
poorly resolved 1H signals. All four proline Cα resonances were
identified above 60 ppm in the HSQC–TOCSY, and each
showed connectivities to Hα and Hβ resonances. The remaining
1H assignments for Pro 8 were obtained by close inspection of
the TOCSY.
Oxaldie-4 exhibited in excess of 100 NOEs between aliphatic
protons and protons in the NH/aromatic region that did not
overlay with TOCSY cross-peaks (Fig. 3A). The NH–NH
region of the NOESY spectrum for Oxaldie-4 showed strong,
clearly resolved sequential NOE cross peaks (Fig. 3B). Clearly
resolved dNN(i,i ϩ 1) NOE cross-peaks were observed. In add-
ition, all medium range dαN(i,i ϩ 3) NOEs were evident in the
segment 14–28 indicative of stable α-helix formation in the C-
terminal region (Fig. 4). These non-sequential NOEs were
absent from residues 1–13, although four residues in this N-
terminal region were prolines lacking the NH proton. For each
of the four proline residues strong dδα(i,i Ϫ 1) NOEs were evi-
dent indicating that these proline amide bonds were in the
trans-conformation. Several long range NOEs were observed.
For example, the side chain of Tyr 27 could be correlated with
the side chains of Pro 2, Glu 4, Pro 5 and Glu 6. Strong NOEs
were also observed between Tyr 20 and Pro 5, Glu 6 and Pro 8
and between the side chains of Pro 5 and Leu 24. These NOEs
between residues of the poly-proline helix and aromatic or
Experimental
Peptide synthesis, purification and identification
Oxaldie-4 was synthesised on Fmoc-5-(4-aminomethyl-3,5-
dimethoxyphenoxy)valeric acid (PAL) on a polyethylene glycol
grafted polystyrene support using a Pioneer Perceptive Biosys-
tems automated peptide synthesiser and standard Fmoc proto-
cols. The following protected amino acids were used: Fmoc-l-
Asn (Tr), Fmoc-L-Thr (But), Fmoc-l-Gln (Tr), Fmoc-l-Asp
(OBut), Fmoc-l-Arg (Pbf ), Fmoc-l-Lys (Boc), Fmoc-l-Ser
(But), Fmoc-l-Glu (OBut), Fmoc-l-Tyr (But). The N-terminal
amine was reacted with acetic anhydride after peptide assembly
and prior to peptide cleavage.
The peptide was cleaved from the polymer and deprotected in
2 ml of 88% TFA, 5% water, 5% phenol and 2% triisopropylsi-
lane per 0.2 g of resin for 2 hours at room temperature. The
J. Chem. Soc., Perkin Trans. 2, 2002, 751–755
753

 Taylor, Susan E.
Taylor, Susan E.